
足细胞衰老与肾小球疾病
2023-11-02 05:59吴其晶综述杨倩倩审校
肾脏病与透析肾移植杂志 2023年5期
吴其晶 综述 杨倩倩 审校
[作者单位]南京医科大学附属淮安第一人民医院肾脏科(淮安,223300)
“世界人口前景”调查显示,至2050年,预计65岁以上人群占总人口的1/6,80岁以上人群的数量将从1.43亿增至4.26亿[1]。随着人口老龄化的加重,衰老相关性疾病带来的经济负担显著增加。其中,衰老相关性肾脏疾病成为一个重要的老年医学问题[2]。35岁以后肾小球滤过率每十年下降约10%[3]。在肾脏组织学,这种变化表现为功能性肾单位的减少,包括足细胞丢失、肾小球硬化、肾小管萎缩、微血管稀疏、间质纤维化及残余肾单位的代偿。
衰老导致肾小球内细胞在数量、结构和功能上发生改变,导致肾脏出现年龄依赖性的肾小球硬化及肾小球滤过率下降。越来越多证据指出,足细胞在肾脏衰老中至关重要。Floege等[4]首次提出衰老的肾小球硬化是“足细胞病”的概念,并已被广泛证实。临床上,衰老导致的肾小球硬化往往伴随足细胞数量和密度的减少,导致移植肾寿命缩短以及老年患者在肾小球疾病中预后更差。作为终末分化细胞,足细胞对衰老的影响高度敏感。近年来,越来越多的研究发现,足细胞衰老是肾损伤的重要环节和潜在治疗靶点[5-7]。因此阐明足细胞衰老在肾损伤中的作用及机制尤为重要。
足细胞衰老的表现
足细胞脱落大量研究表明,足细胞数目与肾小球硬化程度直接相关。随着年龄增长,肾小球硬化率逐渐增多,同时足细胞数目和密度逐渐减少[8]。研究显示,与3月龄小鼠相比,24月龄老年鼠的足细胞密度在皮质和近髓质分别减少45.6%和39%[9]。这些结果在人体得到证实。Wiggins等[10]报道健康成年人每个肾小球足细胞数目在589±166个,该结果与Puelles等[11]报道的558个接近。然而,在70~80岁老年人群中,足细胞储备从年轻肾脏的每个肾小球300个以上下降到100个以下,每年足细胞丢失约0.9%[12]。这种年龄依赖性足细胞丢失具有较高的临床意义,是健康老年肾脏肾小球硬化风险的最佳预测因素。此外,足细胞数量和完整性在移植肾中至关重要。与正常密度的肾移植相比,对于高龄供肾,足细胞数目较少的肾移植发生移植性肾小球病的概率更高[13]。衰老导致足细胞脱落的机制涉及细胞凋亡及焦亡增加,细胞之间的紧密连接减弱等。足细胞需要形成高度复杂的细胞骨架结构,细胞内的肌动蛋白不足以形成纺锤体结构。因此,作为终末分化细胞,足细胞内细胞周期抑制因子高度表达,从而抑制足细胞增殖,衰老进一步促进相关基因表达,抑制足细胞有丝分裂过程中纺锤体形成,从而导致DNA损伤,加重足细胞脱落[14]。
足细胞肥大正常的肾小球需要足细胞覆盖基膜。成年肾小球体积较儿童增加高达700%,每年增加2.7%[11]。同时,足细胞数目也在增加,速度为每年1.8%。衰老引起足细胞减少,而肾小球体积继续增大,最终导致足细胞密度降低[12]。作为终末分化细胞,足细胞不能增殖,为了覆盖裸露的基膜,足细胞出现代偿性肥大,即蛋白质与DNA比值的增加。
Wiggins等[15]等研究表明,为了覆盖增大的肾小球,老化的足细胞肥大过程包括5个阶段。第1阶段为正常足细胞状态;第2阶段为非应激性肥大,即足细胞功能正常;第3阶段为适应性肥大,尽管足细胞内存在应激,但是足细胞功能保持正常;第4阶段表现为失代偿性肥大,足细胞开始丧失功能,同时细胞内存在应激;第5阶段,足细胞肥大不能覆盖肥大的肾小球,导致相对和绝对的足细胞缺失。随着时间推移,这种代偿性肥大不能满足肾脏功能需求,最终导致肾小球硬化。已有研究证实裸露的基膜与瘢痕形成密切相关[16]。与足细胞不同,肾小球其他两种细胞即内皮和系膜细胞在肾小球体积增大的情况下出现增殖。目前,衰老导致的足细胞肥大机制研究较少。其中,p21、p27、转化生长因子β(TGF-β)、哺乳动物雷帕霉素靶蛋白(mTOR)及白细胞介素6(IL-6)可能参与其中[17]。
足细胞基因表达谱改变细胞特异性基因是指在特定细胞类型中表达,决定其形态和功能特异性所必需的一类基因。足细胞含有许多特异性基因,其中裂孔隔膜是维持滤过膜大小和电荷屏障功能正常的一组特定蛋白复合物。包括特异性的膜蛋白,如nephrin 和 podocin;典型的黏附蛋白,如cadherins 和 catenins;支架蛋白,如ZO-1、CD2AP和MAGI-2[18]。这些蛋白水平表达下降导致肌动蛋白细胞骨架的信号传导受损,引起足突异常、蛋白尿并参与肾小球硬化的发生。研究显示,在衰老的足细胞中检测到裂孔隔膜蛋白表达的下调。Wiggins等[15]研究显示,24月龄大鼠肾脏中Tcf21、Nphs1、Col4a5、Glepp1及Wt1表达显著下调。同样,老年老鼠足细胞关键裂孔隔膜、紧密连接和其他特异性足细胞基因和蛋白表达下降[17]。随着足细胞衰老,这些基因的表达改变影响其结构与功能。
肾小球固有细胞相互交流以维持其功能的完整性。Wang等[17]对小鼠的RNA-seq的数据荟萃分析发现,足细胞中特异性富集55对配体-受体相互作用,其中26对受体配体在衰老足细胞中显著减少。这些数据表明,足细胞衰老表现为自分泌和旁分泌信号异常或活性降低,如血管内皮生长因子(VEGF)信号通路,这可能是老年大鼠肾小球内皮细胞数量减少的重要原因。
足细胞衰老的机制
线粒体功能异常、氧化应激及代谢改变线粒体通过产能维持细胞稳态,其功能障碍往往导致活性氧(ROS)增加,而ROS是多种类型细胞衰老的标志之一。研究显示,24月龄小鼠足细胞有广泛的线粒体损伤,包括线粒体内外膜破裂、嵴的缺失、线粒体基质改变和ROS标志物Nox4表达增加[19]。老年小鼠足细胞转录组学研究显示,参与线粒体氧化磷酸化(OXPHOS)的基因包括线粒体呼吸链复合物I-Ⅳ整体下调,ROS下游基因表达上调。本课题组研究结果发现,足细胞线粒体内膜蛋白UCP2特异性敲除小鼠出现年龄依赖性蛋白尿及足细胞损伤(未发表资料),提示线粒体功能异常在足细胞衰老中发挥重要作用。
衰老足细胞的关键代谢途径包括OXPHOS、柠檬酸循环、脂肪酸氧化和氨基酸分解代谢减少[17]。其中,OXPHOS受到广泛关注。其中心调节因子-过氧化物酶体增殖物激活受体共激活因子1α (PGC-1α)在老年足细胞中显著下调。非诺贝特是一种过氧化物增殖物激活受体α(PPARα)激动剂,激活SIRT1和AMPK,促进PGC-1α 表达增加,导致线粒体合成增多。研究显示,给予18月龄老年鼠非诺贝特干预6月后,老年鼠尿蛋白及肾小球硬化减轻,线粒体功能显著改善[20]。但是,足细胞衰老在此过程中是否改善需要进一步探索。最新的研究显示,足细胞能量代谢方式主要是无氧糖酵解而不是OXPHOS[21]。衰老足细胞OXPHOS产能减少,同时伴有FAO基因下调,这种情况下是否会导致衰老足细胞更加依赖糖酵解产能需要进一步研究。综上所述,能量来源和代谢的改变伴随ROS的增加可能加重衰老足细胞的损伤。
SIRT家族表达和活性不足去乙酰化酶(Sirtuins,Sirt)家族通过蛋白质的乙酰化修饰参与自噬等多种细胞生物学过程。其中,Sirt1和Sirt6在足细胞中发挥重要作用。Sirt1表达下调促进足细胞衰老,2017年,Chuang 等[5]发现在足细胞中特异性抑制Sirt1基因,可以增加衰老小鼠肾小球PGC-1α、叉头转录因子O3(FOXO3)、FOXO4和核因子κB(NF-κB)的乙酰化水平,加重了衰老导致的足细胞丢失、微量蛋白尿和肾小球硬化。Huang等[22]研究发现,肾脏Sirt6敲除小鼠在7月龄出现肾小球肥大及蛋白尿,但是,Sirt6在足细胞衰老中的作用还未被报道。分离老年及年轻小鼠原代足细胞进行转录组学分析发现,所有Sirt家族中只有Sirt7出现年龄依赖性表达上调(1.9倍)[17]。并且,Sirt7全身敲除小鼠出现多脏器衰老[23],但是,Sirt7在足细胞衰老中的作用需要进一步研究。
衰老相关分泌表型(SASP)和炎症激活细胞衰老是多种因素导致的细胞周期停滞,端粒缩短及大分子累积。细胞通过诱导衰老的方式不被巨噬细胞清除,从而对抗凋亡,最终这些衰老的细胞在组织中聚集。但是,衰老细胞的慢性积累对器官是有损伤的,很大程度上与这些细胞释放包含SASP相关的蛋白质有关。有研究表明,老年小鼠p16INK4a阳性的衰老细胞显著增多,而特异性敲除p16INK4a,老年小鼠年龄相关性肾小球硬化和肾脏损伤显著改善[24]。Wang等[17]对衰老足细胞进行RNA-seq研究确定了许多衰老相关基因,其中包括11个SASP基因。研究发现衰老足细胞炎症相关基因和炎症小体通路显著激活,其中Nfkb1和Nfkb2在衰老足细胞中表达增加并已被证实参与肾脏衰老[17,25]。
自噬功能异常细胞通过自噬维持蛋白质和细胞器的合成、降解和再循环之间的动态平衡,最终目的是为细胞的生存提供营养物质。作为终末分化细胞,基础状态下,足细胞具有较高的自噬活性。当自噬功能异常时,足细胞会出现类似衰老的表型,同时,自噬功能异常增加了肾脏对FSGS等足细胞病的敏感性。在足细胞中特异性敲除自噬相关基因(Atg5),小鼠足细胞出现衰老加速现象,表现为足细胞数目减少、受损细胞器和异常蛋白的累积[7]。
mTOR复合物是抑制自噬活性的关键分子。研究显示,老年小鼠足细胞mTOR活性显著增强。衰老足细胞中,抑制mTOR活性的基因Prkaa2和Pkhd1表达显著下调[26],而激活mTOR信号通路的基因Cardl1上调近6.9倍[27],提示mTOR参与足细胞衰老。但是,给予老年小鼠雷帕霉素抑制mTOR活性并未显著改善足细胞脱落[28]。因此,在衰老足细胞中,mTOR非依赖性自噬作用需要进一步研究。
表观遗传性改变调控细胞DNA甲基化水平是延缓肾脏衰老的重要策略。多项研究表明,DNA甲基化及转录组改变与肾脏衰老密切相关。其中,Klotho 启动子甲基化是慢性肾脏病的重要特征之一,并且参与肾脏衰老发生发展,大黄抑制Klotho甲基化能显著改善肾脏纤维化及肾损伤[29]。衰老往往伴随细胞内ROS水平升高及端粒反转录酶(TERT)活性下降。研究显示,ROS促进TERT由细胞核向细胞质转移,从而激活Src激酶,最终导致内皮细胞衰老,给予抗氧化剂N-乙酰半胱氨酸能显著能显著抑制ROS产生,抑制TERT向细胞质转移,改善内皮细胞衰老[29]。PPAR激动剂能够缓解衰老导致的肾脏损伤。其机制包括两方面,一是调节p66SHC磷酸化水平,影响线粒体功能改善衰老;其次,PPAR激动剂增加Klotho表达,降低肾脏氧化应激,调节与肾脏衰老相关通路[29]。总体而言,衰老的表观遗传学改变对机体寿命具有重要的意义,并且在生命早期和晚期作用不同,提示与衰老相关的表观遗传学改变可能受到环境因素的影响(图1)。
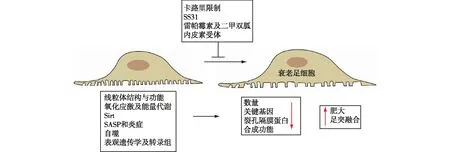
图1 足细胞衰老示意图
延缓足细胞衰老的策略和药物
卡路里限制限制热量摄入是延缓啮齿类动物衰老最常见的方法。Wiggins等[15]研究显示,限制卡路里摄入可预防大鼠年龄相关的肾脏足细胞肥大。同样,Zhang等[30]限制F344老年大鼠卡路里摄入显著缓解足细胞脱落。目前,卡路里限制改善足细胞衰老的确切机制尚不清楚,Sirt可能是其中机制之一[31]。
SS31SS31是一种细胞通透性合成肽,目前正处于治疗心力衰竭、原发性线粒体肌病和其他线粒体疾病的临床试验阶段。SS31通过细胞色素C增强电子传递,增强ATP合成,减少ROS生成,改善线粒体功能[32]。为了研究SS31在足细胞衰老中的作用,Sweetwyne等用SS31干预24月龄小鼠,结果显示,和对照组比较,SS31干预显著改善足细胞线粒体结构异常,抑制ROS生成酶NOX4的表达,增强编码线粒体电子传递链相关酶的基因表达。同时,SS31显著改善老年小鼠足细胞肥大及足突融合,抑制足细胞损伤标志物的表达[19]。
雷帕霉素及二甲双胍mTOR通路调节细胞多种生物学功能,包括翻译、转录和自噬。抑制mTOR诱导自噬,从而在营养剥夺条件下维持细胞存活。研究表明,足细胞中mTOR信号的增加会引起疾病和增加损伤,而在足细胞中特异性减少mTOR信号则会限制损伤。但如上所述,雷帕霉素治疗对老年小鼠足细胞密度没有影响。然而,值得注意的是,在足细胞衰老过程中抑制mTOR信号可以逆转足细胞肥大[33]。二甲双胍是一种口服降糖药,可以延长糖尿病患者寿命。其抗衰老的作用主要通过抑制IGF-1信号发挥降血糖作用。研究显示,二甲双胍能够激活AMPK抑制mTOR活性从而抑制足细胞凋亡,改善糖尿病大鼠足细胞损伤[34]。
内皮素受体darusentan是一种选择性内皮素ETA受体拮抗剂,临床上用来治疗充血性心力衰竭和高血压。有趣的是,darusentan能显著改善23月龄雄性Wistar大鼠足细胞结构,同时,p21和基质金属蛋白酶9(MMP9)的表达减少,但是对小鼠血压或肾素水平无明显影响[35](图1)。
小结:足细胞衰老是肾损伤的重要环节和潜在治疗靶点,探索足细胞衰老形成的核心机制,不仅有助于深入认识老年化过程中多种急慢性肾脏疾病的病理机制,而且对设计新的延缓衰老、阻断疾病进展的综合防治方案有重大临床意义,本文从多维度阐明足细胞衰老过程中的表现、可能参与机制,以及目前针对足细胞衰老提出的预防和治疗策略,尽管已取得一定进展,但应用到临床仍需进一步探索。
猜你喜欢
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
中国医疗保险(2017年5期)2017-05-17
中外医疗(2016年15期)2016-12-01
三峡大学学报(自然科学版)(2016年6期)2016-04-16
中国康复理论与实践(2015年10期)2015-12-24
现代电生理学杂志(2015年1期)2015-07-18
医学研究杂志(2015年9期)2015-07-01
癌变·畸变·突变(2014年1期)2014-03-01
河南医学研究(2014年1期)2014-02-27
