
柱前衍生超高效液相色谱法测量脂蛋白相关磷脂酶A2含量
2023-11-01 08:43:06崔宏恩叶人元朱茂林樊晓琳
中国计量大学学报 2023年3期
崔宏恩,冯 速,叶人元,刘 阳,方 帅,朱茂林,樊晓琳
(1.江苏省计量科学研究院,江苏 南京 210023;2.南京诺唯赞生物科技股份有限公司,江苏 南京 210000)
脂蛋白相关磷脂酶A2(Lp-PLA2)是磷脂酶A2超家族成员,由单核细胞、巨噬细胞或T细胞分泌,Lp-PLA2由441个氨基酸残基组成,相对分子质量为45.4 ku。Lp-PLA2进入血液循环后主要与低密度脂蛋白结合,可产生促炎分子如溶血磷脂酰胆碱和氧化游离脂肪酸[1-3]。Lp-PLA2是血管内皮炎症的特异性指标,其在血液中的含量能够反映动脉粥样硬化的严重程度,能够独立预测经皮冠状动脉介入治疗(PCI)术后冠心病及不良心血管事件(MACE)的风险,从而降低心脑血管患者或高危人群突发心梗/脑梗等严重不良事件的发生概率[4-7]。
目前测定Lp-PLA2的方法有很多种,可因无统一的校准品,所以各种方法测定的结果没有统一的判断标准,对临床应用造成一定的影响[8]。根据ISO17511:2020要求[9],为了保证Lp-PLA2测定结果的准确可比,必须建立可溯源的Lp-PLA2纯品含量测定方法。目前,纯蛋白的定量方法主要有氨基酸分析、质量平衡、定量核磁和同位素稀释质谱法[10-11]。氨基酸分析涉及的多种分析方法在生物化学、蛋白质组学、临床医学、食品科学、化工等领域有着广泛的应用[12]。本文通过对基于柱前衍生的氨基酸分析方法进行优化和改进[13-14],以可溯源到SI单位的氨基酸混合溶液国家标准物质为标准,以满足临床检验的需求。
本文首先采用SDS-PAGE和质谱对Lp-PLA2纯品进行纯度表征、相对分子质量测定和蛋白质鉴定,然后对Lp-PLA2酸水解、柱前衍生化和色谱条件等进行优化,建立起Lp-PLA2纯品蛋白含量的柱前衍生超高效液相色谱(UPLC)测定方法。另外,以高效液相色谱-同位素稀释质谱(HPLC-IDMS)方法(目前公认用于蛋白含量测定的基准方法)测定结果作为参照进行对比,考察所建立柱前衍生UPLC方法的方法学参数。
1 仪器与试剂
1.1 仪器
超高效液相色谱仪(Acquity,美国Waters公司),配自动进样器和紫外检测器;超高效液相色谱仪(EASY-nLC 1200,美国Thermo公司);质谱仪(Orbitrap Fusion Lumos Tribrid,美国Thermo公司);电泳仪(美国Bio-Rad公司);天平(ME235S,分度值0.01 mg,德国Satorius公司);常温高速离心机(pico7,美国Thermo公司);pH计(FE28,美国METTLER TOLEDO公司)移液器(10 μL,100 μL,200 μL,1 000 μL,美国Thermo公司);超纯水系统(GOKU-BI,厦门锐思捷水纯化技术有限公司);集热式恒温加热搅拌器(DF-101S,上海予华仪器有限公司);超声波清洗机(XO-5200DTS—10L-300W,南京先欧仪器制造有限公司);循环水真空泵(SHZ-D(III),上海予华仪器有限公司)。
1.2 试剂
氨基酸混合溶液标准物质(GBW(E)100062,中国计量科学研究院);6-氨基喹啉基-N-琥珀酰-亚胺基甲酸酯(AQC)衍生剂(上海阿拉丁生化科技股份有限公司);三氟乙酸(色谱纯,美国Sigma-Aldirich公司);甲酸(色谱纯,德国Fluka公司);乙腈(色谱纯,美国Sigma-Aldirich公司);蛋白质电泳预制胶(12%,常州天地人和生物科技有限公司);10×SDS缓冲溶液(南京诺唯赞生物科技股份有限公司);彩虹预染宽分子量蛋白Marker(14-99 ku,日本TAKARA公司);胰蛋白酶(美国Promega公司);二硫苏糖醇(DTT,美国Sigma-Aldirich公司);碘乙酰胺(瑞典Amersham公司);碳酸氢铵(IAM,美国Sigma-Aldirich公司);天冬酰胺粉末、谷氨酰胺粉末、色氨酸粉末、半胱氨酸粉末(美国Sigma-Aldirich公司);无水乙酸钠(色谱纯,美国Sigma-Aldirich公司);三乙胺(色谱纯,南京化学工业公司);乙二胺四乙酸二钠(色谱纯,美国Sigma-Aldirich公司);硼酸(色谱纯,美国Sigma-Aldirich公司);三氯乙酸(色谱纯,北京国药集团化学试剂有限公司);醋酸(色谱纯,上海沪试试剂有限公司);氢氧化钠(色谱纯,北京国药集团化学试剂有限公司);盐酸(色谱纯,南京化学工业公司);甲醇(色谱纯,美国TEDIA公司);γ-氨基丁酸(色谱纯,上海阿拉丁生化科技股份有限公司);10 kd过滤离心管(德国Satorius公司),其余试剂均为分析纯。
2 Lp-PLA2基本性质表征
2.1 SDS-PAGE纯度测定
将Lp-PLA2样品(以南京诺唯赞生物科技有限公司提供的人源重组Lp-PLA2纯品为原料分装于合适的管中)与5×loading buffer(含β-巯基乙醇)在一个1.5 mL离心管中按4∶1的体积比混合。混匀后盖紧管盖。在电磁炉沸水中煮沸5 min,完全冷却后瞬时离心5 s。用10 μL移液器吸取样品进行上样,在相应加样孔加入5 μL相对分子质量蛋白Marker。将电压调至80~100 V,待样品中溴酚蓝指示带电泳至分离胶位置(约200 min);再以160~200 V电压将溴酚蓝指示带电泳至距玻板最下方约5 mm处停止电泳,时间约30 min。分析结束后用考马斯亮蓝染色1 h时,加脱色液(水、甲醇和冰醋酸以5∶4∶1的比例配制)过夜脱色。
2.2 相对分子质量测定和蛋白质鉴定
2.2.1 样本前处理
取离心后的Lp-PLA2上清样品加入到10 kd超滤离心管中,10 000 r/min离心20 min;而后加入5 mmol/L DTT常温反应25 min;加入0.5 mol/L IAM至终浓度为14 mmol/L,避光反应30 min后再次10 000 r/min离心20 min;加入100 μL的50 mmol/L碳酸氢铵后10 000 r/min离心20 min;更换新的套管,滤膜上加入含有1 μg胰酶的100 μL的50 mmol/L碳酸氢铵,37 ℃酶解16~18 h。过夜酶解的肽段离心加酸终止酶解,用C18柱除盐后真空冷冻干燥。
2.2.2 液相色谱-质谱联用分析[15]
取肽段粉末,使用体积分数0.1%甲酸复溶后使用EASY-nLC 1200超高效液相仪进行洗脱分析。流动相A为体积分数0.1%甲酸水溶液;流动相B为含0.1%甲酸的乙腈溶液。洗脱梯度:3%~5%B,5 s;5%~15%B,40 min;15%~28%B,34 min 50 s;28%~38%B,12 min;38%B~100%B,5 s;100%B,8 min;流速300 nL/min。肽段经洗脱电离后串联至Orbitrap Fusion Lumos Tribrid质谱进行分析。离子源电压设置为2.2 kV,肽段母离子及其二级碎片都使用高分辨的Orbitrap进行检测和分析。一级质谱扫描范围设置为350~1 500m/z,扫描分辨率设置为60 000;二级扫描分辨率设置为15 000。数据采集模式使用数据依赖型扫描DDA,HCD碰撞池使用30%的碎裂能量进行碎裂,串联质谱扫描的动态排除时间设置为30 s,相对分子质量平行测定6次。
2.2.3 数据库检索
二级质谱数据使用Proteome Discoverer(v2.4)进行检索。检索参数设置:数据库使用Uniprot-homo-sapiens-proteome_UP000005640和uniprot_mouse_proteome_UP000000589(2021.04),酶切方式设置为Trypsin/P;漏切位点数设为2;肽段最小长度设置为6个氨基酸残基;将半胱氨酸烷基化设置为固定修饰,将甲硫氨酸的氧化,蛋白N端的乙酰化作为可变修饰。
3 柱前衍生-UPLC测定Lp-PLA2蛋白含量[16]
3.1 Lp-PLA2样品的水解
向待测Lp-PLA2样品加入6 mol/L的盐酸,使用氮吹仪排尽空气后密封,在(110.0±0.5)℃的烘箱中进行水解,通过对水解时间进行优化后,选取96 h作为该样品最优水解时间,取出平衡至室温后通过氮气吹干其中的盐酸,然后加入1 000 μL 0.01 mol/L的盐酸复溶。
3.2 配制内标工作液
经过优化,选择γ-氨基丁酸作为内标,以确保样品峰附近没有杂质峰的出现,不会对样品峰形成干扰,且和样品分离完全,保留时间相差不大。首先,称取10 g三氯乙酸,溶解于100 mL超纯水中形成10%三氯乙酸溶液。然后,称取2.062 4 mgγ-氨基丁酸,使用100 mL10%三氯乙酸溶液溶解成200 μmol/Lγ-氨基丁酸溶液,即内标工作液。
3.3 配制混合标准工作液
首先,分别使用0.1 mol/L盐酸将天冬酰胺、谷氨酰胺、色氨酸、半胱氨酸配制成10 mmol/L单标储备液(通过优化,配制单标储备液,是为了便于观察其他氨基酸是否会干扰17种氨基酸)置于2~8 ℃保存,有效期3个月。然后,吸取17种氨基酸混合溶液国家标准物质及配置的4种单标储备液,用超纯水稀释成0.5 mmol/L的21种混标氨基酸标准工作液500 μL置于2 ℃~8 ℃保存,有效期1周。将21种混标氨基酸标准工作液稀释成氨基酸浓度分别为0.02 mmol/L,0.05 mmol/L,0.08 mmol/L,0.10 mmol/L,0.30 mmol/L,0.50 mmol/L的混合标准工作液。
3.4 水解样品的衍生化
首先,分别吸取各待测Lp-PLA2水解样品、混合标准工作液200 μL至1.5 mL离心管中。其次,吸取内标工作液200 μL至以上各离心管中。再次,将各离心管于8 000~10 000 r/min转速离心10 min。最后,吸取各离心管离心后的上清液10 μL、70 μL硼酸缓冲液、20 μL AQC衍生溶液(经过比较和优化,以AQC作为柱前衍生剂),于55 ℃水浴10 min。溶液过0.22 μm滤膜后入进样小瓶待测。
3.5 UPLC分析
经优化,确定如下色谱条件:色谱柱YMC-Triart C18 250×4.6 mm S-5 μm,12 nm,流动相A为17 mmol/L三乙胺(pH=5.65)+140 mmol/L乙酸钠+1 mg/L乙二胺四乙酸二钠溶液;流动相B为乙腈,柱温为30 ℃,进样体积为5 μL,检测波长为248 nm,流速为1.0 mL/min。UPLC流动相梯度洗脱表如表1。
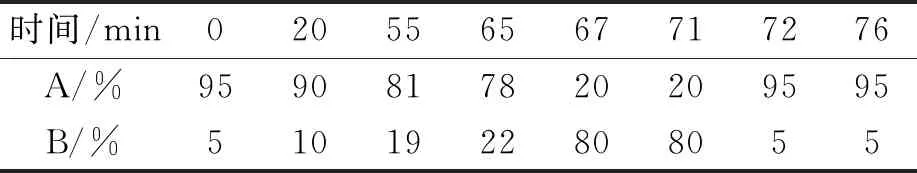
表1 UPLC流动相梯度洗脱表
采用UPLC依次对配制的混合标准工作液和Lp-PLA2样品溶液进行检测,记录UPLC信号强度,利用混标氨基酸标准工作液建立氨基酸浓度-峰面积标准曲线,计算Lp-PLA2样品中各氨基酸浓度。
3.6 结果计算
根据如下公式计算待测蛋白的质量浓度:
ρR=cA×MR/n。
(1)
其中:ρR为待测蛋白样品的质量浓度,mg/mL;cA为所选择氨基酸的物质的量浓度,mmol/L;MR为待测蛋白样品的相对分子质量;n为待测样品每个分子中所选择氨基酸的个数。
所选择氨基酸为天冬氨酸、异亮氨酸、苏氨酸、亮氨酸、丝氨酸、酪氨酸、谷氨酸、苯丙氨酸、甘氨酸、赖氨酸、丙氨酸、组氨酸、胱氨酸、精氨酸、缬氨酸、脯氨酸和甲硫氨酸中的任一种或任几种,所选择氨基酸为任几种时,待测样品中蛋白的质量浓度为每种氨基酸计算出的待测样品中蛋白的质量浓度的平均值。计算时选择的氨基酸种类越多,计算蛋白含量的平均值误差就越小,实际可以根据情况进一步选择17种氨基酸中分离洗脱情况比较好的氨基酸种类进行计算。
4 结果与讨论
4.1 Lp-PLA2基本性质的表征
4.1.1 SDS-PAGE测定结果
纯品Lp-PLA2的SDS-PAGE电泳测定结果见图1,Lp-PLA2条带位于45.4 ku附近,与构建的蛋白相对分子质量理论值48.615 35 ku和文献[1]、[2]报道均一致。
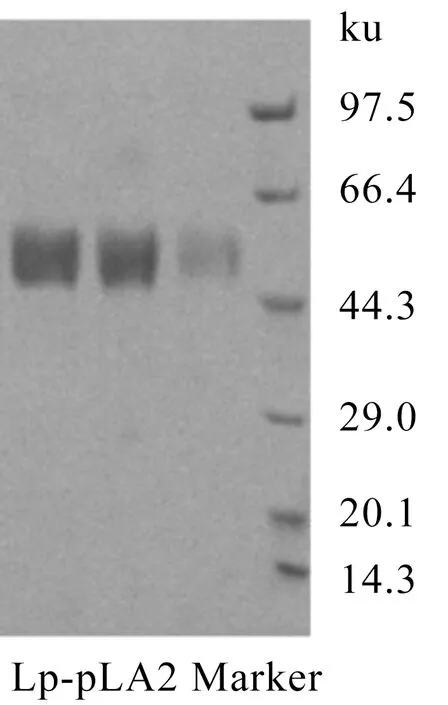
图1 纯品Lp-PLA2的SDS-PAGE电泳纯度测定结果Figure 1 SDS-PAGE electrophoresis purity determination results of pure Lp-PLA2
4.1.2 相对分子质量测定和蛋白质鉴定结果
纯品Lp-PLA2的相对分子质量测定结果和酶切后的质谱图分别见图2和图3。结合图2和图3,由于Lp-PLA2相对分子质量相对较大,离子化效率较差,不易去卷积,未得出完整相对分子质量,后续含量计算时以构建的蛋白相对分子质量理论值48.615 35 ku为准。图3中可见酶切出的肽段,采用Proteome Discoverer(v2.4)软件在Uniprot-homo-sapiens-proteome_UP000005640和uniprot_mouse_proteome_UP000000589(2021.04)数据库中使用酶切后的质谱数据进行蛋白质鉴定,结果表明,所鉴定肽段来自于Lp-PLA2,得分为323.31。
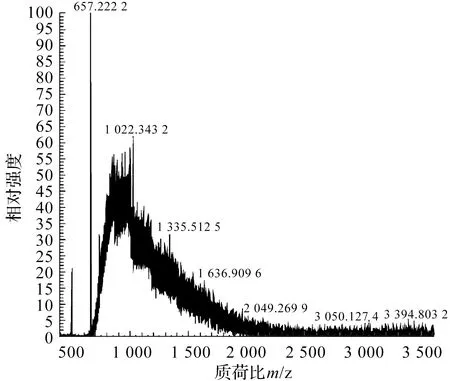
图2 纯品Lp-PLA2的相对分子质量测定结果Figure 2 Relative molecular weight determination of pure Lp-PLA2

图3 纯品Lp-PLA2酶切后的质谱图Figure 3 Mass spectrum after enzymatic digestion of pure Lp-PLA2
4.2 柱前衍生UPLC法评价[10]
本研究以可溯源到SI单位的GBW(E)100062氨基酸混合溶液标准物质为标准,建立了Lp-PLA2纯品蛋白含量的柱前衍生UPLC测定方法。首先,对柱前衍生UPLC法测定纯品Lp-PLA2结果的不确定度进行评定。实验采用了丝氨酸(Ser)、丙氨酸(Ala)、异亮氨酸(Ile)和赖氨酸(Lys)对蛋白质含量分别进行测定,并取其平均值,各氨基酸浓度的计算式表示为
cA=Ax×c0/A0。
(2)
式(2)中:cA为待测蛋白样品中所选择氨基酸的物质的量浓度,mmol/L;Ax为待测蛋白样品中所选择氨基酸的色谱峰面积,μV·s;c0为标准工作液中所选择氨基酸的物质的量浓度,mmol/L;A0为标准工作液中所选择氨基酸的色谱峰面积,μV·s。
因此应分别计算各个氨基酸对蛋白质定量结果的不确定度,以Ser为例,测量不确定度的主要来源为称量、定容、色谱峰面积、水解效率、方法重复性和氨基酸混合溶液标准物质[10]。根据天平的检定证书及规程JJG 1036-2022确定称量的不确定度[17],称量10 g三氯乙酸、2.062 4 mgγ-氨基丁酸量、氨基酸混合溶液标准物质(250 mg)及配置的4种单标储备液(25 mg)时的最大允许误差均为0.015 mg,按照均匀分布,计算得到称量引入的相对不确定度分量分别为
um=
0.426%。
配制内标工作液和混合标准工作液定容过程中,使用100 mL单标线A级容量瓶2次,10 mL单标线A级容量瓶4次,根据JJG 196—2006《常用玻璃量器检定规程》[18],100 mL单标线A级容量瓶的最大允许误差为±0.10 mL,10 mL单标线A级容量瓶的最大允许误差为±0.020 mL,按照三角分布;实验室温度为(20±5)℃,水的体积膨胀系数为2.1×10-4/℃,按照均匀分布,则由定容引入的相对不确定度分量uV为
uV=
0.184%。
根据仪器说明书和积分仪的一般性能指标分析,目前用于仪器色谱峰面积积分处理的最大误差为0.2%~1%,取1%[19],按照均匀分布,则色谱峰面积引入的相对不确定度分量uAx/A0为
按照不确定度A类评定方法评价该方法重复性引入的不确定度分量,6次重复测定结果的RSD为2.48%,因此,测定结果平均值的重复性相对不确定度分量ur为
根据GBW(E)100062氨基酸混合溶液标准物质证书,其中Ser的标准值为0.994 mmol/L,U=0.03 mmol/L(k=2),所以由Ser标准物质引入的相对不确定度分量uSer为
水解效率引入的相对标准不确定度ue均按照1%估算[10],则采用Ser测定蛋白质含量结果的标准合成相对不确定度为
uc,Ser=
同样地,采用Ala、Ile和Lys测定蛋白质含量结果的标准合成相对不确定度分别为
uc,Ala=2.28%,uc,Ile=1.98%,uc,Lys=1.98%。
取k=2,最后结果的扩展不确定度为
U=2uc=
方法的扩展不确定度为
uk=0.118 3 mg/mL×U=0.002 6 mg/mL。
采用优化建立的柱前衍生UPLC方法测定同一Lp-PLA2样品,重复测定6次,所得结果的平均值为0.118 3 mg/mL,RSD为2.48%,可见所建方法重复性良好。采用优化建立的柱前衍生UPLC方法测定6个Lp-PLA2样品,每个样品重复测定3次,所得结果的平均值为0.118 1 mg/mL,RSD为2.41%,可见所建方法再现性良好。
4.3 与纯品Lp-PLA2含量HPLC-IDMS测定结果的对比分析
取6个纯品Lp-PLA2样品,委托中国计量科学研究院采用HPLC-IDMS对每个样品重复测定3次,实验采用了缬氨酸(Val)、亮氨酸(Leu)、异亮氨酸(Ile)和苯丙氨酸(Phe)对蛋白质含量分别进行测定,并取其平均值。经计算,HPLC-IDMS测定的纯品Lp-PLA2含量为0.119 4 mg/mL,RSD为4.52%。同时,参照柱前衍生UPLC法不确定度评定思路(不确定度的主要来源为称量、水解效率、方法重复性和氨基酸标准物质)计算的HPLC-IDMS方法的扩展不确定度为1.7%(k=2)。与HPLC-IDMS测定结果相比纯品Lp-PLA2的柱前衍生UPLC法测定结果具有-0.93%的相对偏差,在柱前衍生UPLC法测定结果的不确定度范围内。同时,柱前衍生UPLC法与HPLC-IDMS法之间的归一化偏差( En)值为0.33,说明柱前衍生UPLC法对纯品Lp-PLA2的测定结果相对准确可靠,以其作为一种补充方法用于纯品Lp-PLA2标准物质定量是可行的。
4.4 柱前衍生UPLC方法的检出限与定量限评价
柱前衍生UPLC方法的检出限和定量限为
其中:DLOD为检出限,mg/mL;DLOQ为定量限,mg/mL;cA为标准曲线最小浓度样品中所选择氨基酸的物质的量浓度,mmol/L;MR为待测蛋白样品的相对分子质量;n为待测样品每个分子中所选择氨基酸的个数;N为噪音;S为检测器灵敏度。
经过计算,Lp-PLA2的柱前衍生UPLC法的检出限与定量限分别为1.96×10-2mg/mL和6.55×10-2mg/mL。
5 结 语
本文主要建立了纯品Lp-PLA2含量的柱前衍生UPLC测定方法,经过方法学参数考察和与HPLC-IDMS测定结果的比较表明,所建立的柱前衍生UPLC方法溯源清晰、准确度高,相比HPLC-IDMS方法成本更低,更便于推广应用,可作为一种补充验证方法用于纯品Lp-PLA2标准物质的定值,在建立我国Lp-PLA2测定结果的量值溯源传递体系、保证相关试剂盒生产和临床诊断中Lp-PLA2测定结果的准确可比具有重要意义。
猜你喜欢
色谱(2022年11期)2022-11-10 03:36:42
色谱(2022年10期)2022-10-13 12:42:40
色谱(2022年7期)2022-06-25 01:54:28
色谱(2022年4期)2022-04-01 01:42:24
中国洗涤用品工业(2019年4期)2019-05-11 09:27:18
中成药(2018年1期)2018-02-02 07:20:05
制造技术与机床(2017年9期)2017-11-27 02:14:23
动物医学进展(2015年10期)2015-12-07 05:46:19
西南石油大学学报(自然科学版)(2015年3期)2015-04-16 05:12:08
食品工业科技(2014年7期)2014-03-11 18:15:07
