
衰老相关分泌表型相关的lncRNAs 模型预测肝细胞癌患者预后的构建和验证
2023-10-28 00:50周丁杰曹德东
中国医药导报 2023年25期
周丁杰 戈 伟 曹德东
武汉大学人民医院肿瘤中心,湖北武汉 430060
肝细胞癌(hepatocellular carcinoma,HCC)是导致全球癌症死亡的第四大原因[1],每年造成80 多万人死亡。HCC 患者的5 年生存率仅为5%~30%[2]。尽管目前靶向治疗和免疫疗法已进入临床,但肝癌患者的预后仍然较差。寻找新的治疗靶点及检测预后生物标志物仍是当下研究的重点。衰老相关分泌表型(senescence-associated secretory phenotype,SASP)是指由衰老细胞分泌的细胞因子、生长因子和酶等分子,这些分子可引发慢性炎症反应,促进肿瘤的形成和发展[3]。研究表明,SASP 在肝癌中的作用与其成分有关。例如,SASP 中的白细胞介素(interleukin,IL)-6、IL-8 等因子被认为是肝癌的重要促进因子,能够促进肝癌细胞的增殖和侵袭,抑制免疫系统的反应[4];而转化生长因子β 等因子则能够促进肝癌干细胞的增殖和转移[5]。在肝癌中,SASP 参与了肿瘤微环境的形成和转化过程[6],并且lncRNA 作为SASP 中的重要组成部分,也在肝癌中发挥了重要作用。研究显示,GAS5 参与了肝癌中的SASP,可以通过抑制IL-6 的表达来抑制肿瘤细胞的增殖和侵袭,并提高肝癌患者的生存率[7]。另外,GAS5 还可以通过调节核因子-κB 信号通路来抑制肝癌细胞的转移和侵袭[8]。lncRNA-H19 可以通过介导细胞内活性氧水平的升高,促进IL-6 和IL-8 等SASP 相关因子的分泌,从而促进肝癌细胞的增殖和侵袭[9]。上述结果显示,SASP 相关的lncRNA 有望成为肝癌诊断和治疗的新靶点。基于SASP 相关的lncRNA,本研究建立了一个包含6个lncRNA 的风险模型,旨在优化肝癌临床治疗,改善肝癌患者预后[9-16]。
1 材料与方法
1.1 实验材料
在癌症基因组学图谱(the cancer genome stlas,TCGA)数据库[10]中检索并下载了424 例HCC 患者样本的RNA 测序数据,临床资料包括年龄、性别、生存时间、生存状态、临床分期及TNM 分期,排除有模糊值和临床资料不完整以及随访时间<30 d 的样本后,最终纳入241 例HCC 样本。并按1∶1 的比率将纳入样本按计算机随机分为实验组(120 例)和验证组(121例)。通过Ensembl 网站[11](http://asia.ensem-bl.org/)对转录组数据注释以区分mRNA 和lncRNA 数据,提取HCC 的lncRNA 表达矩阵。
1.2 实验方法
1.2.1 差异表达基因及lncRNA 的筛选 在GeneCards网站[12](https://www.genecards.org/)中获取相关系数>7 的201 个SASP 基因,随后利用R 软件中的“limma”包[13]对HCC 样本和正常样本中的SASP 相关基因进行差异分析,筛选标准为|logFC|≥1 且FDR<0.05,得到差异基因。随后设定相关系数为|r|>0.4 且P<0.001,对HCC 样本中的lncRNAs 和差异基因进行Spearman 相关分析,获取SASP 相关lncRNAs。
1.2.2 预后风险模型的建立 利用Perl 语言脚本将临床数据和对应的lncRNA 矩阵样本对应后合并。使用R 软件中“survival”包(https://github.com/therneau/survival)对SASP 相关lncRNAs 进行单因素Cox 回归分析,筛选出HCC 预后相关的lncRNAs。采用“glmnet”软件包[14]进行的lasso 回归分析对筛选出的lncRNA 进一步限制,采用十倍交叉验证来获得lambda 惩罚参数的理想值。经过lasso 回归解决共线性问题,再经过多因素Cox 回归分析后,将得到的lncRNAs 纳入风险模型中。lncRNA 模型的计算公式为:风险得分=(coefi×xi)。系数值用“Coef”表示,所选lncRNAs 的表达值用“x”表示。
1.2.3 风险模型的评估 应用风险模型计算出每个HCC 患者的风险分数。根据风险评分的中位数,将HCC 患者分为低风险组和高风险组。对高风险组和低风险组用Kaplan-Meier 曲线评估生存差异,绘制受试者操作特征(receiver operating characteristic,ROC)曲线评估风险模型灵敏度与特异度。单因素和多因素Cox回归分析比较风险模型和其他临床病理变量(年龄、性别、临床分期、TNM 分期)的关系,运用R 软件“ggpubr”包(https://rpkgs.datanovia.com/ggpubr/)绘制了不同临床病理参数下的高、低风险组之间的生存差异。
1.2.4 风险模型的免疫图谱分析 利用Perl 语言将矩阵与含风险值的样本文件合并,得到高、低风险组中不同样本基因的表达量新矩阵。使用“GSVA”工具[15]探索样本的风险评分与样本中免疫细胞浸润和经典的免疫检查点相关标志物之间的关系。
1.3 统计学方法
所有分析皆由R 软件(4.1.3)和Perl 完成。散点图展示样本的风险评分与生存状态的关系,生存曲线采用Kaplan-Meier(K-M)法生成,使用单因素和多因素Cox 比例风险回归模型评估SASP 相关lncRNAs预后模型,采用ROC 曲线评价预测模型的可靠性和准确性。计量资料采用t 检验,计数资料采用χ2检验或Fisher确切概率法。以P<0.05 为差异有统计学意义。
2 结果
2.1 HCC 中SASP 相关差异表达基因(differential gene expression,DEGs)及lncRNA 的筛选
本研究对TCGA 数据库中的HCC 队列的RNA测序数据清洗后共纳入241 例样本。通过“limma”包对HCC 样本和正常样本进行差异分析,得到58 个显著差异的SASP 基因。其中有36 个上调基因及22 个下调基因(图1A)。热图展示了所有的DEGs 在样本中的表达情况(图1B)。并利用Spearman 分析得到DEGs 的1 048 个lncRNAs。
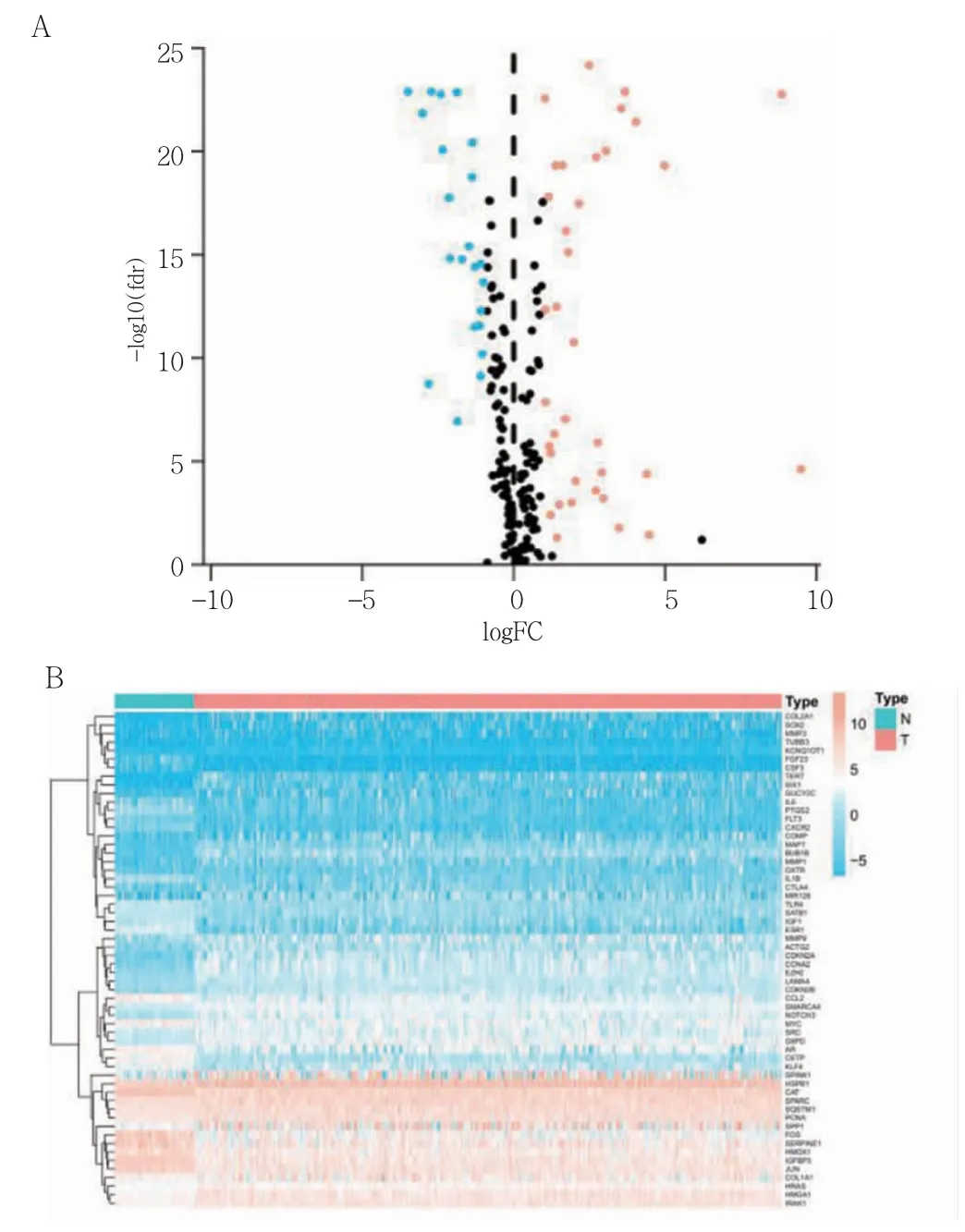
图1 肿瘤组织和正常组织间的DEGs 表达
2.2 构建lncRNA 风险模型
利用Perl 脚本从测序数据中分离出SASP 相关lncRNA 的表达矩阵,随后联合生存资料,利用单因素Cox 回归分析确定了64 个预后相关且差异表达的lncRNAs(P<0.001)。随后对这些lncRNAs 进行了Lasso 回归,得到14 个SASP 相关的lncRNAs(图2A),通过10 轮交叉验证确定了最佳惩罚参数值(图2B)。最后通过多因素Cox 回归分析,从上述14 个lncRNAs中筛选出6 个lncRNAs,包括AL031985.3、AC124067.4、AC009283.1、AC107021.2、LINC00324、HPN-AS1,构成风险模型。公式:风险评分=AL031985.3 expression×(0.768 307)+AC124067.4 expression×(0.301 32+AC009 283.1 expression×(-0.367 63)+AC107021.2 expression×(0.495 064)+LINC00324 expression×(-0.511 08)+HPNAS1 expression×(-0.751 87)。多因素Cox 回归分析结果见表1。图2C 展示了模型中6 个lncRNAs 在HCC样本中的表达情况。本研究使用Cytoscape 来进一步可视化lncRNAs 和对应的mRNA 的关系(图2D,|R2|>0.4 和P<0.001)。Sankey 图显示,AC009283.1、HPN-AS1 和LINC00324 是保护因素,而AC107021.2、AC124067.4 和AL031985.3 是风险因素(图2E)。
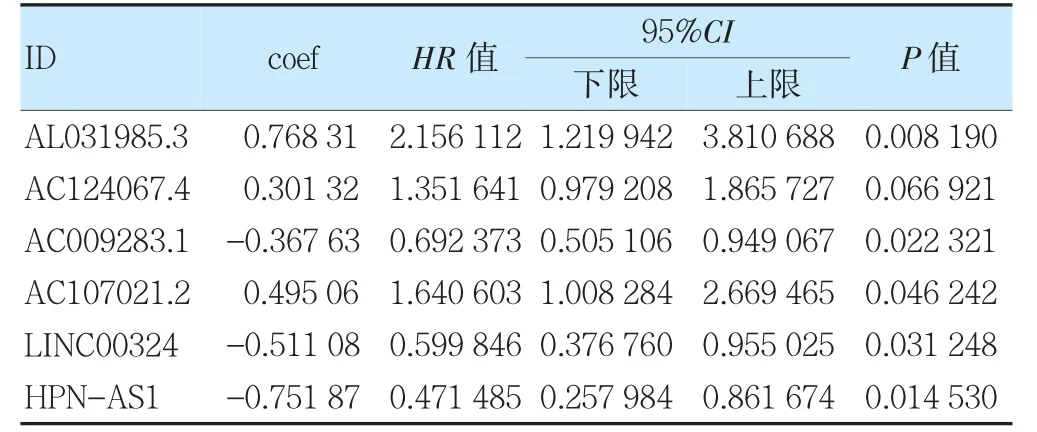
表1 多因素Cox 回归分析筛选用于构建预后风险模型的SASP 相关lncRNA
2.3 风险模型的评估
2.3.1 高、低风险组间的生存分析 生存分析结果显示,HCC 患者的预后随着风险值升高而恶化(图3A)。K-M 曲线结果显示,与高风险组比较,低风险组患者的预后更好(P<0.001)(图3B)。风险值ROC 曲线显示,1、3、5 年生存率的曲线下面积(AUC)分别为0.812、0.754、0.739(图3C)。
2.3.2 单因素和多因素Cox 回归分析 为了进一步评估此风险模型在HCC 中的应用,本研究结合了其他病理参数,包括年龄、性别、临床分期、TNM 分期,在实验组和验证组中分别进行单因素和多因素Cox 回归分析。结果显示,在实验组中,风险评分的HR 在单变量Cox回归分析中为1.437(95%CI:1.280,1.614)(图4A,P<0.001)。在多因素Cox 回归分析中,HR 值为1.409(95%CI:1.242,1.599)(图4B,P<0.001)。在实验队列中,单变量、多变量Cox 的风险评分HR 值分别为1.372(95%CI:1.209,1.557)(图4C,P<0.001)和1.366(95%CI:1.178,1.585)(图4D,P<0.001),提示此风险模型可作为HCC 患者的独立预后因素。
猜你喜欢
中国宝玉石(2019年5期)2019-11-16
天津医科大学学报(2019年3期)2019-08-13
知识经济·中国直销(2018年8期)2018-08-23
数学学习与研究(2017年3期)2017-03-09
冰雪运动(2016年4期)2016-04-16
中国老区建设(2016年1期)2016-02-28
肿瘤预防与治疗(2015年1期)2015-09-26
中国当代医药(2015年16期)2015-03-01
癌变·畸变·突变(2015年4期)2015-02-27
军事历史(1981年2期)1981-08-14
