
DPAGT1基因突变致先天性肌无力综合征1例报告并文献复习▲
2023-10-19 13:25刘自强赖兰娣
广西医学 2023年15期
刘自强 赖兰娣 刘 芬
(1 东莞市儿童医院康复医学科,广东省东莞市 523326; 2 中山大学附属东华医院预防保健科,广东省东莞市 523129; 3 东莞市儿童医院小儿神经内科,广东省东莞市 523326)
先天性肌无力综合征(congenital myasthenic syndrome,CMS)是一组由于遗传缺陷导致的神经肌肉接头形成、维持与功能缺陷的罕见遗传性疾病,主要表现为波动性肌肉无力、易疲劳等,可能导致呼吸暂停而危及生命,常于新生儿或婴儿期发病[1]。神经肌肉接头的正常运作依赖突触前膜、突触间隙和突触后膜的众多蛋白相互协调来共同完成信号传递,糖基化修饰在这一过程中必不可少。乙酰胆碱受体糖基化缺陷会导致终板膜乙酰胆碱受体的缺失,进而降低突触后膜对乙酰胆碱的反应性,临床上表现为四肢肌肉无力、喂养困难、运动发育迟滞等[2]。目前,已发现有30多个基因突变与CMS的发病有关,大多数为常染色体隐性遗传,而常染色体显性遗传主要为慢通道综合征[3]。本研究分析1例由于多萜醇磷酸-N-乙酰氨基葡萄糖磷酸转移酶(dolichyl-phosphate N-acetylglucosamine-phosphotransferase 1,DPAGT1)基因突变导致CMS的患儿的临床特点及遗传学特征,以提高临床医生对该病的认识。
1 临床资料
患儿男性,6月龄,因“运动发育迟缓、四肢肌张力低下”于2019年3月1日至东莞市儿童医院小儿神经内科门诊就诊,然后于2019年3月5日至小儿神经内科办理住院。患儿出生后喂养正常,3月龄时出现抬头不稳,可吸吮手;6月龄时视觉和听力发育尚可,仍抬头不稳,不能翻身、独坐,精神状态不佳,活动后2 h左右即睡觉。患儿系第1胎第1产,出生胎龄为39+2周,阴道分娩出生,出生体重2 700 g,出生时无窒息、羊水吸入史,其父母身体健康、非近亲结婚,否认遗传病家族史。入院时体格检查:神志清楚,精神可,头围40 cm,体重5.5 kg,身长61 cm,前囟大小为2 cm×2 cm,平坦,视听反应尚可,追踪不持久,头可竖立,但不稳;拉起反射头背屈,俯卧位可抬头50 °,但不能持久,不能翻身;四肢肌张力明显降低,内收肌角150 °、腘窝角150 °、足背屈角60 °,双上肢肱二头肌、肱三头肌、双侧膝反射、跟腱反射减弱,四肢肌力4+级,双侧巴氏征阴性。入院时辅助检查:甲状腺功能正常;头颅MRI和脑电图检查未见明显异常;血串联质谱、尿有机酸分析未见异常;肌电图显示,所检左腓总神经、左尺神经重复频率刺激(+),提示神经肌肉接头损害;乙酰胆碱受体抗体阴性。基因检测结果提示,患儿存在DPAGT1基因(转录本号:NM_001382)的复合杂合突变c.284T>C(p.Phe95Ser)和c.976C>T(p.Leu326Phe),测序数据显示这两个突变分别遗传自其父亲和母亲(均为杂合状态)。这两个突变在参考人群基因数据库中分别为没有报告和频率较低,同时经初步生物信息学分析显示,这两个位点在不同物种间高度保守(见图1A)。蛋白质3D结构预测结果显示,第95位氨基酸由疏水性的苯丙氨酸(Phe)变为亲水性的丝氨酸(Ser),与周围氨基酸残基之间的氢键连接没有明显改变,但是该位点位于跨膜结构区域,推测其会影响蛋白的功能;与野生型比较,第326位氨基酸由亮氨酸(Leu)变为Phe后,Phe与周围残基之间的氢键连接发生了改变(见图1B),提示该位点为致病性突变的可能性大。
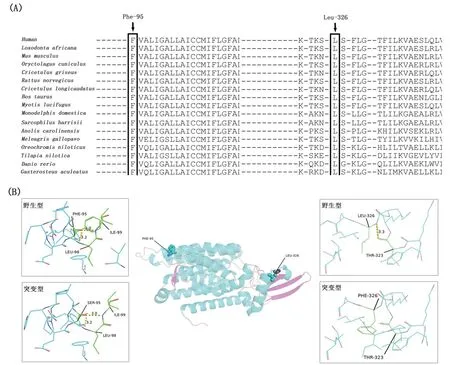
图1 DPAGT1基因突变c.284T>C(p.Phe95Ser)和c.976C>T(p.Leu326Phe)的软件预测结果
由于入院初期未行肌电图及基因检测,临床诊断考虑为“松软儿”,给予康复运动训练、鼠神经营养药物治疗2周,疗效不佳。完善肌电图及基因检测后,结合患儿临床特点,予修正诊断为CMS(DPAGT1基因突变)。因此停用鼠神经营养药物治疗,给予口服溴吡斯的明片,15 mg/次,3次/d。服药治疗2周后,患儿活动耐力较前改善,生命体征平稳,予办理出院。出院后继续予口服溴吡斯的明(15 mg/次,3次/d)治疗,服药2个月后患儿运动功能较前好转,耐力明显增加,易疲倦感明显减轻。患儿2岁时返回东莞市儿童医院小儿神经内科门诊复诊,体格检查发现患儿精神可,理解能力和反应能力可,能简单对答,可主动用言语表达需求,可独自行走500 m,步态略不稳,偶有跌倒,四肢腱反射可引出,双侧巴氏征阴性。
2 讨 论
CMS为罕见的神经肌肉接头疾病[4],神经肌肉接头依赖突触前膜、突触间隙和突触后膜的众多蛋白相互协调来共同完成信号传递,其中任何相关基因突变均可能影响神经肌肉接头功能,导致CMS[5]。目前主要根据突变基因相关蛋白在神经肌肉接头处的解剖分布和功能对CMS进行分类,包括突触前膜蛋白缺陷、突触间隙蛋白缺陷、突触后膜蛋白缺陷、终板发育与维持缺陷、糖基化缺陷和其他罕见类型[4]。CMS临床特点为起病早,常在新生儿或婴幼儿期发病,疾病进展缓慢,表现为眼部、躯干、肢体肌肉无力,常有运动发育迟滞,不耐受疲劳等,使用乙酰胆碱酯酶抑制剂治疗有效[6]。但因基因突变和分子遗传机制的不同,CMS的发病年龄、临床症状、肌无力受累范围及治疗效果各异。本文报告的CMS患儿于婴儿期发病,临床主要表现为四肢肌无力、活动后易疲劳,智力发育基本正常,经基因检测明确为DPAGT1基因复合杂合突变引起的AChR糖基化异常所致。
先天性糖基化异常(congenital disorder of glycosylation,CDG)是一类由于聚糖合成或与其他复合体(蛋白质和脂质)结合过程缺陷引起的先天性代谢疾病,主要是由于人体内蛋白N-糖基化途径缺陷引起。根据酶缺陷发生环节的不同,CDG可分为CDG-Ⅰ和CDG-Ⅱ两种类型,其中脂联寡糖前体合成并转移到新生多肽途径的缺陷被定为CDG-Ⅰ型,DPAGT1基因突变引起的亚型则为CDG-Ⅱ型[7-9]。
DPAGT1基因编码多萜醇磷酸-N-乙酰氨基葡萄糖磷酸转移酶1,在脂联寡糖生物合成的第一步,多萜醇磷酸-N-乙酰氨基葡萄糖磷酸转移酶1催化N-乙酰葡萄糖胺,使N-乙酰葡萄糖胺转移到位于内质网膜中的多萜醇磷酸,合成多萜醇焦磷酸N-乙酰氨基葡萄糖[10]。因此,DPAGT1-CDG是N-糖基化过程中的催化酶DPAGT1基因突变导致的,DPAGT1蛋白缺乏会导致天冬酰胺糖基化受损,从而出现DPAGT1-CDG相关的一系列临床表现,主要包括先天性肌无力、难治性癫痫、发育迟缓和智力障碍等[4]。若患者临床上仅表现为肌无力而无神经肌肉以外其他系统受累,则称为DPAGT1-CMS,此类患者肌肉选择性受累的原因尚不明确,部分学者认为是由于DPAGT1基因突变造成乙酰胆碱受体糖基化缺陷而引起终板膜乙酰胆碱受体缺失,进而引起突触后膜乙酰胆碱的反应性下降所致[2]。Engel[4]对1988年至2017年梅奥诊所诊断的359例CMS患者进行统计分析,发现糖基化缺陷CMS患者占3.63%。
目前已有13例DPAGT1基因突变相关的CMS患者被报告[11-15](见表1),CMS患者多起病于婴幼儿期,临床表现为全身肌无力,多为近端肌无力[16],易疲劳,随年龄增长病情趋于稳定或逐渐进展,大多数患者均可存活。大部分CMS患者认知功能正常,但部分DPAGT1-CMS患者有轻度认知功能障碍或学习能力低下[17],目前认为其认知功能障碍可能与中枢神经系统受累有关。有学者发现,已报告确诊为DPAGT1-CMS的13例患者中有5例存在学习障碍或智力障碍,1例为自闭症患者,部分DPAGT1-CMS患者肌肉活检显示管状聚集物,该基因突变所致的CMS对溴吡斯的明治疗均敏感[11-15]。
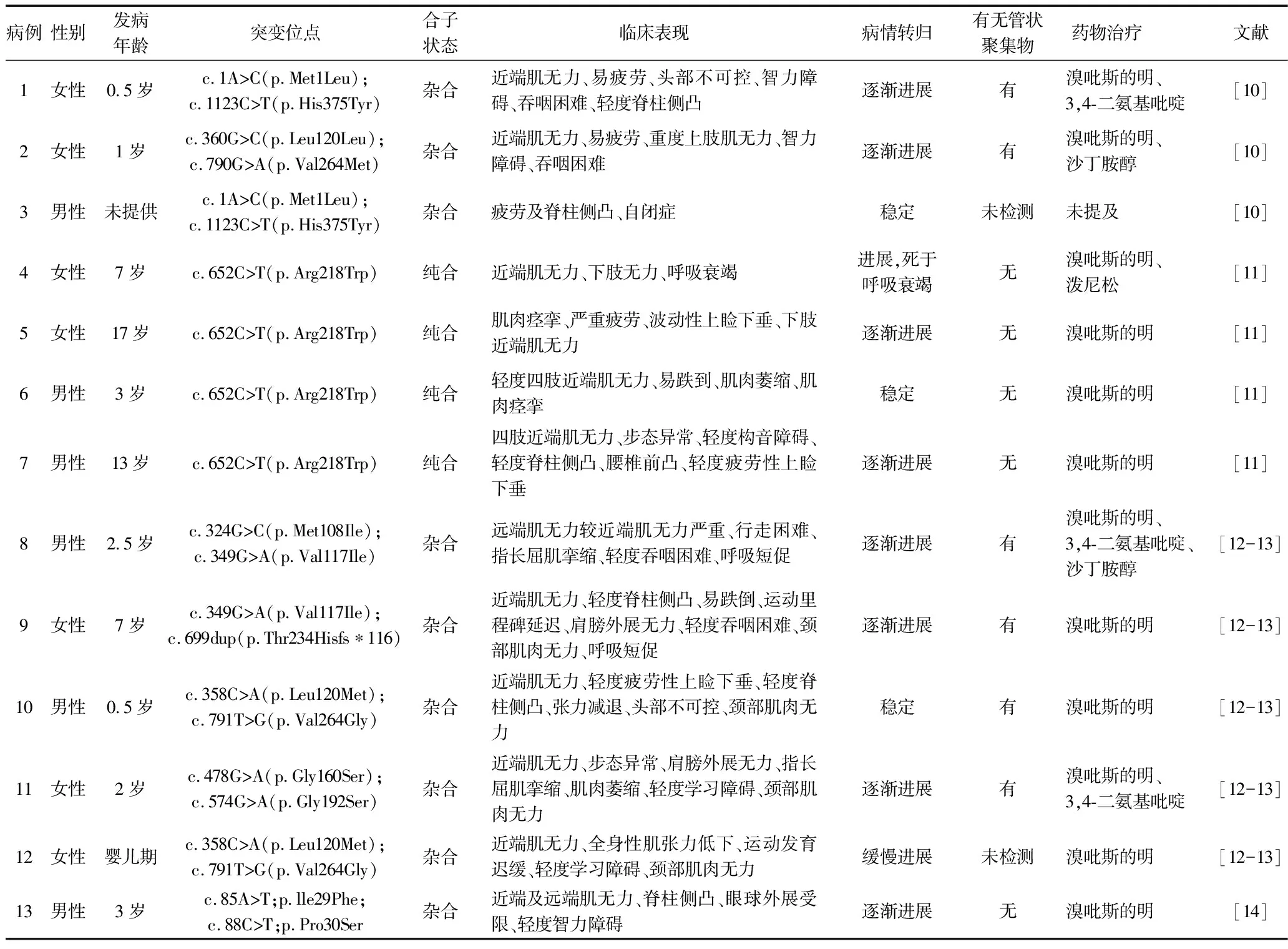
表1 13例DPAGT1基因变异相关的GMS患者的临床特点
本研究检测出该例患儿DPAGT1基因存在两个新的错义突变c.284T>C(p.Phe95Ser)和c.976C>T(p.Leu326Phe),这两个突变位点分别来源于其父亲和母亲,且目前该两个突变类型尚无文献报告。这两个突变类型在人群基因数据库中出现的频率较低,且在不同物种间高度保守,蛋白质3D结构预测结果显示,第326位的Leu变为Phe后,影响Leu跟周围氨基酸残基之间的氢键连接,结合患儿肌无力和易疲劳的临床特点,提示DPAGT1基因突变是导致该患儿出现CMS的遗传学病因,诊断明确后给予口服溴吡斯的明治疗2个月后,患儿耐力明显改善。随访患儿至2岁,发现其肌无力症状明显改善,能独自行走,步态略不稳,智力发育尚可,表明溴吡斯的明治疗DPAGT1-CMS效果较好,与既往文献报告相似[1]。
3 小 结
CMS的临床表现和遗传异质性较大,临床上不易与其他神经肌肉疾病(如肢带型肌营养不良)相鉴别,容易被误诊或漏诊。在临床上遇到肌无力症状、起病年龄早且康复治疗效果欠佳的肌无力患者,应考虑CMS的可能,可尽早行遗传学检测以明确诊断并进行针对性治疗。此外,除慢通道综合征、乙酰胆碱酯酶缺乏症等少数亚型外,多数CMS患者应用溴吡斯的明治疗效果良好,可明显改善患者预后,可作为一线治疗方案。系统学习并掌握CMS的临床特点和遗传学特征,结合基因检测明确诊断和分型,对于CMS患者的治疗和预后至关重要。
猜你喜欢
现代畜牧科技(2021年6期)2021-07-16
中国民间疗法(2021年5期)2021-06-09
世界科学技术-中医药现代化(2020年2期)2020-07-25
国际放射医学核医学杂志(2020年2期)2020-05-30
中国中医急症(2019年10期)2019-05-21
现代检验医学杂志(2016年4期)2016-11-15
医学研究杂志(2015年12期)2015-06-10
中国粮油学报(2014年7期)2014-02-06
现代检验医学杂志(2014年1期)2014-02-06
实用医药杂志(2012年6期)2012-04-21
