
1 例COL4A4 基因5 号外显子c.274G>T:p.G92*无义突变的薄基底膜肾病患者家系遗传分析
2023-10-18 06:37:56李静朱井俊邵坤段子才王飞
山东医药 2023年27期
李静,朱井俊,2,邵坤,段子才,王飞
1 阜阳市第六人民医院检验科,安徽阜阳 236001;2 阜阳市第二人民医院肾脏内科;3 阜阳师范大学体育学院;4 安徽理工大学医学院检验系
薄基底膜肾病,也称为良性家族性血尿,是一种常见的遗传性肾脏疾病,其发病率较高。该疾病是儿童和成人持续性镜下血尿的最常见原因之一,临床特征主要表现为持续性血尿和轻度蛋白尿,通常不会出现眼耳等其他器官的受累,并且预后较好[1-2]。患者通常具有正常的肾功能,其惟一或主要的病理表现是弥漫性肾小球基底膜变薄[2-3]。1996年,LEMMINK 等[1]报道,薄基底膜肾病的主要致病原 因 与 编 码Ⅳ型 胶 原α3、α4 链 的COL4A3 和COL4A4 的基因突变相关,该基因突变导致胶原蛋白结构或数量改变,进而影响基底膜的正常功能和结构稳定性。同时,在其他家系研究中,也报道了编码Ⅳ型胶原α3、α4和α5链的基因COL4A3、COL4A4和COL4A5 可能为致病原因,其中,一些常见的COL4A4 基因突变位点包括c.4001A>G:p.G1334E错义突变位点、c.5044C>G:p.R1682G 错义突变位点和c.871-1G>A:splicing 剪切突变位点等。上述这些结果提示薄基底膜肾病与Alport综合征(AS)有较为密切的关系,具有遗传异质性。一般认为,多数薄基底膜肾病患者不会发展为慢性肾功能衰竭或终末期肾病,但10%~20%的薄基底膜肾病患者会在平均年龄60 岁时发展为终末期肾病[4]。目前,由于没有明确的循证治疗方案可用于薄基底膜肾病,因此,了解COL4A4 基因突变引起的薄基底膜肾病的临床表型特征及遗传特点对于该疾病预后、治疗策略和监测方案的制定非常重要。我们发现1 例由COL4A4 基因5 号外显子c.274G>T:p.G92*无义突变所致的薄基底膜肾病,现将其家系遗传特点报道如下。
1 资料分析
患者女,64岁。于2016年无明显诱因下出现双下肢轻度水肿,活动后水肿症状加重,休息后稍缓解,无腰疼,无肉眼血尿,无胸闷、气喘,未重视。半年后患者因水肿症状加重,呈凹陷性,伴有腰酸、乏力、夜尿增多等不适症状,至当地卫生院就诊。查尿常规提示潜血阳性(+++)、蛋白尿阳性(++),血肌酐203 μmol/L,肾脏彩超提示双肾大小尚可,临床诊断“慢性肾炎、肾功能不全”。为进一步明确病因,就诊于本院,征得患者同意后给予行肾穿刺活检术。肾活检病理提示肾小球基底膜厚度变薄,符合薄基底膜肾病,建议行基因检测进一步明确诊断。经与患者及其家属沟通后,患者未同意行基因检测。给予护肾、排毒、降压、减少尿蛋白等药物治疗延缓肾功能进展,并嘱其注意休息、避免劳累、慎用肾毒性药物,出院后患者没有进行规律门诊复查随访、也未按时服用药物。
2021 年6 月患者因恶心、呕吐、胸闷就诊本院。实验室检查尿常规提示潜血阳性(+++)、尿蛋白阳性(++++),血 肌 酐1 008.8 μmol/L,尿 素 氮23.94 mmol/L,血红蛋白74 g/L,肾脏超声提示双肾缩小、呈慢性损害性声像图。结合薄基底膜肾病史,临床明确诊断为慢性肾衰竭—尿毒症期;薄基底膜肾病。给予维持性血液透析治疗,2~3 次/周,病情总体尚平稳。
同时因为家系中有多人肾脏相关疾病出现,对患者3 代以内的家庭成员进行调查,该家系成员儿子(Ⅱ1、Ⅱ4)、女儿(Ⅱ2、Ⅱ3)均有血尿和蛋白尿,先证者的孙女Ⅲ2、孙子Ⅲ4和外甥女Ⅲ5均有血尿,其他家庭成员尿检无异常。为了进一步明确该病的病因以及是否存在遗传性肾病,征得患者及其家人同意后,我们采集了先证者和其家系成员的外周静脉血2 mL,使用Illumina Hiseq2500 测序仪对其进行全外显子组测序,最后通过Sanger 测序对检测到的变异位点进行了家系验证。
全外显子组测序发现,先证者(Ⅰ1)COL4A4 基因5 号外显子存在c.274G>T:p.G92*无义突变。Sanger 测序结果(见图1)显示该突变遗传符合家系共分离,在患者与家族成员中携带者遵循一致的遗传模式,均符合常染色体显性遗传,提示该突变位点可能是致病位点。先证者儿子(Ⅱ4)、女儿(Ⅱ2、Ⅱ3)、孙女(Ⅲ2)、孙子(Ⅲ4)、外甥女(Ⅲ5)COL4A4 基因5 号外显子均存在c.274G>T:p.G92*无义突变。其中先证者儿子(II1)因外出务工未进行血样采集化验,其女儿(Ⅲ2)和儿子(Ⅲ4)均携带该突变,再结合Ⅱ1 有血尿、蛋白尿症状,推测出家庭成员(Ⅱ1)携带该突变。对检测出的COL4A4 基因突变进行数据库检索,发现其为新发突变位点。
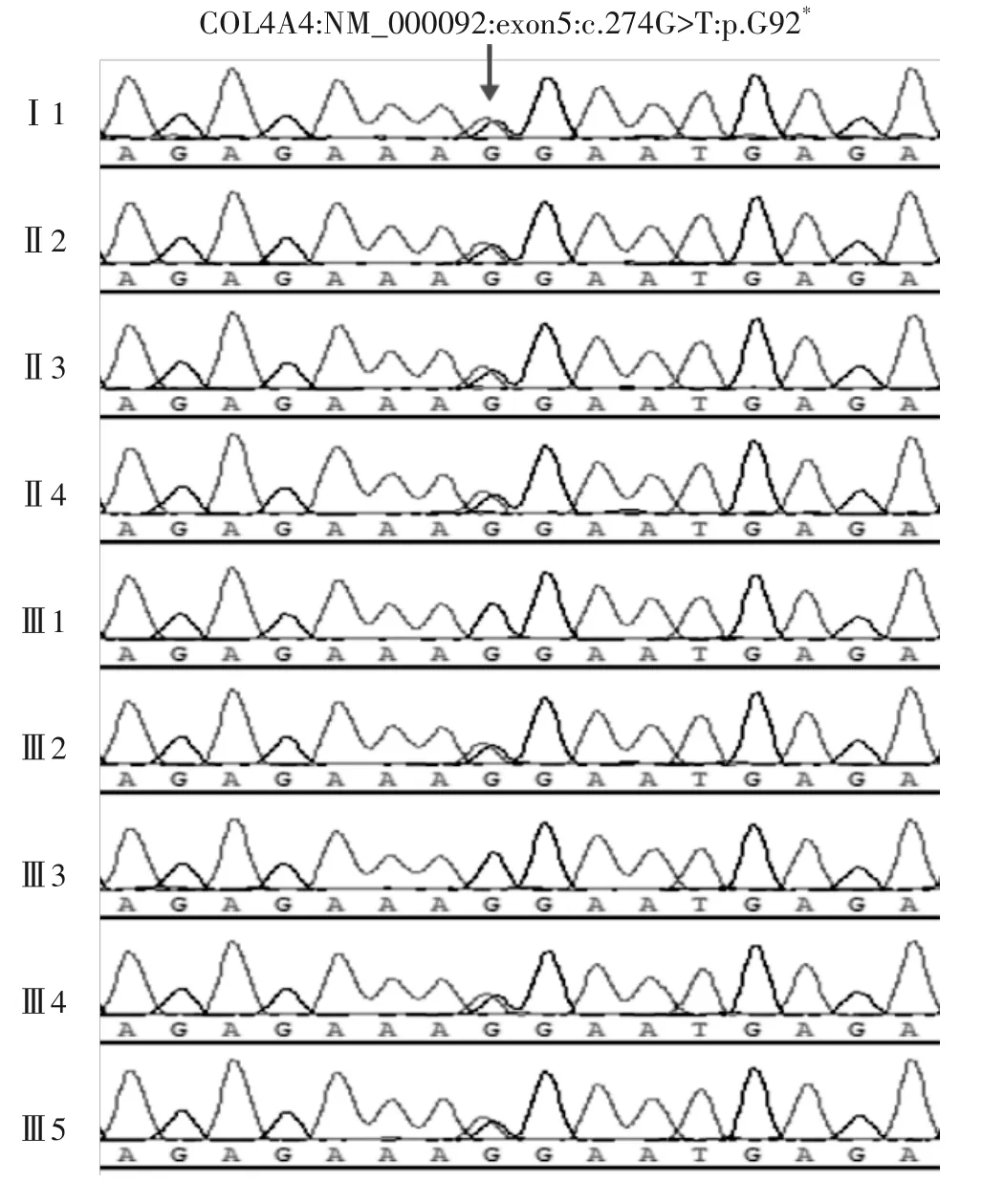
图1 薄基底膜肾病家系中家庭成员COL4A4基因突变位点Sanger测序结果
按照美国医学遗传学与基因组学学会(ACMG)的遗传突变分析指南,对先证者家系成员进行遗传分析,并参考遗传病致病数据库在线人类孟德尔遗传数据库(OMIM)、人类基因突变数据库(HGMD)以及PubMed 等相关文献进行分析。通过综合遗传方式和先证者的突变基因分析确定该遗传病的遗传方式,并鉴定出家系共分离的突变位点。最终根据先证者的临床特征筛选出与先证者临床特征最匹配的致病突变为5号外显子.274G>T:p.G92*突变。
在Uniprot和Ensembl数据库中筛选人类和其他物种COL4A4 基因同源蛋白序列,用DNAman 软件进行蛋白序列同源比对,发现p.G92*位点甘基酸在人类和其他物种相同位置上高度保守(见图2)。应用Swiss-model 同源建模软件对新发现的变异进行蛋白质三维结构同源建模分析比对,变异c.274G>T:p.G92*导致编码产物由甘氨酸替换成终止密码子,与野生型蛋白相比有显著差异(见图3),说明该突变可能会导致COL4A4 蛋白的形态异常和功能障碍。以上结果均显示该突变为薄基底膜肾病的潜在致病突变位点。
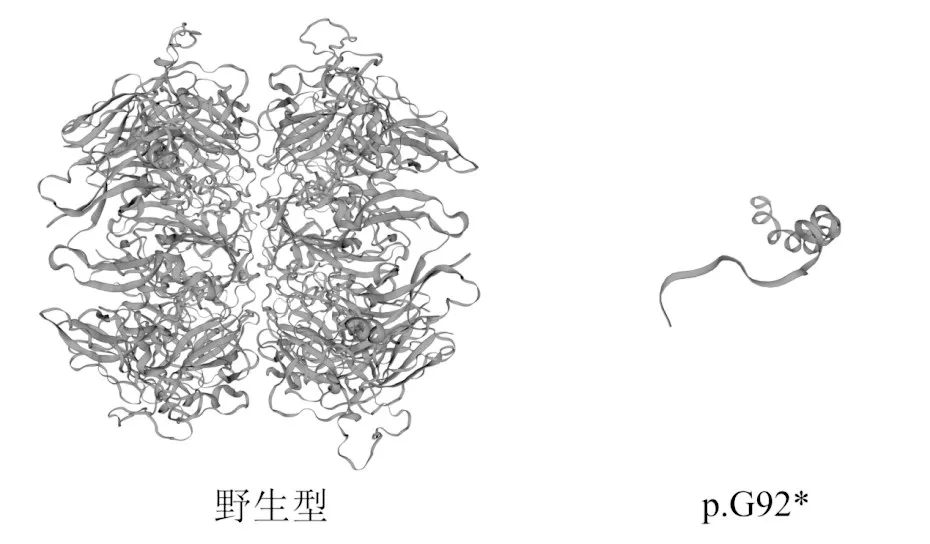
图3 Swiss-model同源建模构建的COL4A4蛋白三维结构
2 讨论
薄基底膜肾病的遗传方式是一种常染色体显性遗传疾病,其发病通常与COL4A3/COL4A4 基因的突变有关。该疾病的主要临床表现是血尿,多数患者表现为持续的镜下血尿,也可能出呼吸道感染或剧烈活动后的发作性肉眼血尿。该疾病可以在任何年龄发生,但多数患者的预后良好[5]。
在GBM 中,Ⅳ型胶原作为含量最丰富的分子之一,它与基底膜中的层黏连蛋白、巢蛋白等相互作用,共同参与组成网络结构[6]。目前已鉴定出六种不同的IV 型胶原蛋白链,分别被命名为α1至α6,这些蛋白链由COL4A1 至COL4A6 基因进行编码。随着肾脏的发育,α1α2 链逐渐被α3α4α5 链取代,因此α 3α4α5链是出生后GBM的主要成分,其交联度更高,更耐蛋白质降解,这可能对肾小球选择性屏障特性的建立和维持至关重要[7]。COL4A3和COL4A4位于2q36.3 上,COL4A3 包含52 个外显子并编码α3 链(1 670 个氨基酸),而COL4A4包含48个外显子并编码α4 链(1 690 个氨基酸)[8]。薄基底膜肾病中COLA43 和COL4A4 突变可能影响阻碍GBM 中α3α4α5交联网络结构的形成,并最终降低GBM 的厚度和稳定性,导致薄基底膜肾病或AS的发生。
AS 的临床表现比薄基底膜肾病复杂,主要表现为肾功能进行性恶化、高频感音神经性耳聋和眼部病变,有些AS患者早期或者不典型的情况下只有血尿一种表现,需要与薄基底膜肾病进行区分。AS的遗传模式有多种,其中X连锁显性遗传型占大多数,约85%,通常由COL4A5 基因发生突变导致,其余15%左右是常染色体显性遗传或隐性遗传,由COL4A3 或COL4A4 基因发生突变引起[9]。当存在持续性肾小球血尿、少量蛋白尿(500 mg/d)、肾功能正常(大部分)且无其他明显肾外异常时,临床上可诊断薄基底膜肾病[10]。大约40%的薄基底膜肾病家庭成员有血尿,血尿与COL4A3/COL4A4 基因座的突变分离,约三分之二的薄基底膜肾病患者中,至少有一名其他家庭成员患有常染色体显性遗传模式的血尿[11]。
以往研究认为薄基底膜肾病是良性的,在健康人群中可出现,其肾脏超声、肾功能、血压等均正常。本研究中,先证者64 岁,病情逐渐发展到加重阶段,并开始进行透析治疗,该家系成员II1、II2(38岁)、II3(36 岁)、II4(35 岁)均有血尿、蛋白尿,病情较先证者轻。先证者孙女III2(12 岁),孙子III4(7岁)以及外甥女III5(13 岁)均有血尿,无其他症状。有研究[12]发现,薄基底膜肾病的症状与年龄有相关性,患者会随着年龄增长而渐渐发展到更加严重的临床阶段,出现蛋白尿、局灶阶段性肾小球硬化、肾功能下降等临床表现,这也表明薄基底膜肾病并不如之前所认为的那样良性。
薄基底膜肾病的遗传模式是常染色体显性遗传,即只要一个等位基因上出现有害的突变,就可能引起疾病的发生。全外显子组测序发现先证者(I1)COL4A4 基因5 号外显子存在c.274G>T:p.G92*无义突变,使92 号甘氨酸突变成终止密码子,进而肾小球基底膜三聚体组装异常,这破坏了Ⅳ型胶原蛋白三螺旋结构的稳定性和柔韧性,最终影响到基底膜网络的功能。该突变经Sanger测序结果证实与家系共分离相符合,在蛋白序列同源比对中发现,该变异位点在人类和其他物种之间具有高度的保守性,利用Swiss-model同源建模软件构建出的COL4A4蛋白野生型和变异型的三维结构,可以清楚地看到变异体p.G92*无法形成完整的蛋白结构,这种无义突变导致翻译过程提前结束,所编码的蛋白质无法正常表达,对蛋白质功能的影响很大。
总之,COL4A4 基因5 号外显子c.274G>T:p.G92*无义突变致薄基底膜肾病临床表现为血尿,病理表现为基底膜变薄,晚期可能会发展为终末期肾病;该突变引起的遗传模式为常染色体显性遗传。根据ACMG 指南,其突变类型可能为致病性突变。此突变位点目前在国内外未见报道,补充了人类薄基底膜肾病基因突变数据库,对进一步研究中国人群薄基底膜肾病的发病机制、遗传咨询等提供了依据。
猜你喜欢
听力学及言语疾病杂志(2022年5期)2022-09-20 09:07:10
临床输血与检验(2022年3期)2022-06-22 02:52:50
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
中国自行车(2018年8期)2018-09-26 06:53:34
振动与冲击(2018年4期)2018-03-05 00:34:24
实用临床医药杂志(2016年21期)2016-12-09 03:28:12
人人健康(2016年21期)2016-11-05 11:05:31
重庆医学(2015年12期)2015-03-05 05:52:54
振动与冲击(2014年23期)2014-05-16 07:01:56
