
高效液相色谱法测定芴甲氧羰基-L- 亮氨酸中的有关物质
2023-10-09 10:36朱凯莉刘虎卞从明董世建程婷李增礼安徽安科生物工程集团股份有限公司
安徽科技 2023年9期
文/朱凯莉 刘虎 卞从明 董世建 程婷 李增礼[安徽安科生物工程(集团)股份有限公司]
多肽药物是指通过生物合成法(天然原料提取法、酶解法、发酵法、基因重组法)或化学合成法(液相合成法和固相合成法)获得的具有特定治疗作用的多肽,具有生物活性高、特异性强、免疫原性低及稳定性强等优点,广泛应用于抗病毒、抗肿瘤、抗菌、疫苗、内分泌、心血管及糖尿病等医疗领域。目前,合成多肽药物应用最普遍的是固相合成法,以保护氨基酸(Boc-氨基酸或Fmoc- 氨基酸)作为起始物料,定向获得目标肽,由于Fmoc- 氨基酸具有在碱性条件下进行脱保护的优势,因此是目前固相合成法中最为常用的起始物料。关于起始物料中杂质的控制,食品药品监督管理局(FDA)、欧洲药品管理局(EMA)以及ICHQ11、《合成多肽药物药学研究技术指导原则》均对原料药(API)起始物料质量控制提出评估意见或要求。芴甲氧羰基氨基酸作为固相合成多肽中的起始物料,引入的杂质及所产生的相关杂质在纯化过程中可能与目标肽共同洗脱,因此杂质很难减少或消除,为了尽量减少对终产物的潜在影响,需对此类杂质建立严格的质控要求。
检测固相合成多肽中起始物料的方法有很多,其中高效液相色谱法(HPLC)具有灵敏度高、分析速度快、重复性好及操作简单等诸多优点。有关物质检测杂质含量低、种类多,故更适合采用HPLC 法进行检测。本文以芴甲氧羰基-L- 亮氨酸(Fmoc-L-Leu-OH)作为研究对象,建立了HPLC 检测Fmoc-L-Leu-OH有关物质的方法,并进行了系统的方法学验证。
一、材料
1.主要材料和试剂
Fmoc-L-Leu-OH 对照品,杂质对照品:N- 羟基琥珀酰亚胺(HOSu)、9- 芴甲氧羰基- 琥珀酰亚胺碳酸酯(Fmoc-OSu)、9- 芴甲醇(Fmoc-OH)、N- 芴甲氧羰基-β- 丙氨酸(Fmoc-β-Ala-OH)、N- 芴甲氧羰基-β- 丙氨酸- 亮氨酸(Fmoc-β-Ala-Leu-OH)、N- 芴甲氧羰基- 亮氨酰- 亮氨酸(Fmoc-Leu-Leu-OH),乙腈,磷酸,注射用水。
2.主要仪器设备
赛默飞U3000 高效液相色谱仪,紫外检测器,电子天平。
二、方法与结果
1.试液配制
系统适用性溶液:分别称取Fmoc-L-Leu-OH 对照品与杂质对照品HOSu、Fmoc-OSu、Fmoc-OH、Fmoc-β-Ala-OH、Fmoc-β-Ala-Leu-OH、Fmoc-Leu-Leu-OH 适量,精密称定,加乙腈制成每1 mL 含Fmoc-Leu-OH 1.0 mg 与杂质 HOSu、Fmoc-OSu、Fmoc-OH、Fmoc-β-Ala-OH、Fmoc-β-Ala-Leu-OH、Fmoc-Leu-Leu-OH 各约3.0 微克的混合溶液作为系统适用性溶液。
定位溶液:精密称取各已知杂质(HOSu、Fmoc-OSu、Fmoc-OH、Fmoc-β-Ala-OH、Fmoc-β-Ala-Leu-OH、Fmoc-Leu-Leu-OH)适量,加乙腈溶解并稀释分别制成浓度约1.0 毫克/毫升的杂质定位溶液;精密称取Fmoc-L-Leu-OH 适量,加乙腈溶解并稀释配制成浓度约为1.0 毫克/毫升的样品溶液。
供试品溶液:取本品适量,精密称定,加乙腈溶解并稀释制成浓度约为1.0 毫克/毫升的溶液,作为供试品溶液。
对照溶液:精密量取供试品溶液适量,加乙腈定量稀释制成每1 毫升中约含Fmoc-Leu-OH 1.0 微克的溶液,作为对照溶液。
以乙腈作为空白溶剂和稀释液。
2.色谱条件
采用Waters XSelect CSHTM C18(4.6×250 mm,5微米)色谱柱,以0.15%磷酸∶乙腈(60∶40)为流动相进行梯度洗脱,洗脱程序如表1 所示,流速为1.0 毫升/分,进样量为5 微升,柱温为35 ℃,检测波长为220纳米。

表1 流动相梯度洗脱程序
3.计算方式
本次方法学验证各杂质采用加校正因子的自身对照法计算,计算公式如下:
已知杂质=AS×f/AR×0.1%
单一未知杂质=Ai/AR×0.1%
总杂质=∑所有杂质
式中:AS为供试品溶液中的已知杂质峰峰面积;Ai为供试品溶液中的未知杂质峰峰面积;AR为对照溶液的峰面积;f为已知杂质的相对校正因子。
4.限度
HOSu、Fmoc-OSu、Fmoc-OH、Fmoc-β-Ala-OH、Fmoc-β-Ala-Leu-OH、Fmoc-Leu-Leu-OH 均不得大于对照溶液主峰面积(0.1%),其他单个杂质不得大于对照溶液主峰面积的2 倍(0.2%),总杂质不得大于对照溶液主峰面积的5 倍(0.5%)。
5.检测波长的确定
取系统适用性溶液的Fmoc-Leu-OH 和已知杂质(HOSu、Fmoc-OSu、Fmoc-OH、Fmoc-β-Ala-OH、Fmoc-β-Ala-Leu-OH、Fmoc-Leu-Leu-OH)在190~400 纳米紫外扫描光谱图,结果发现HOSu 在波长230纳米以上几乎无紫外吸收,流动相中乙腈的截止波长是190 纳米,结合文献研究结果,故将Fmoc-L-Leu-OH 有关物质检测波长定为220 纳米。
6.方法学验证
(1)专属性:取各定位溶液分别进样检测,结果已知杂 质 HOSu、Fmoc-β-Ala-OH、Fmoc-OH、Fmoc-β-Ala-Leu-OH、Fmoc-OSu、Fmoc-Leu-Leu-OH 与Fmoc-L-Leu-OH 的保留时间分别为2.647、8.593、9.163、10.277、11.580、14.022、13.155 分钟。取空白溶剂、适用性溶液进样检测,色谱图如图1 所示。结果空白溶剂对各杂质检测无干扰,主峰与相邻杂质之间的分离度分别为11.59、6.07,杂质峰之间的分离度分别为62.26、4.60、8.78、10.11,分离度远大于2.0,表明方法专属性良好。
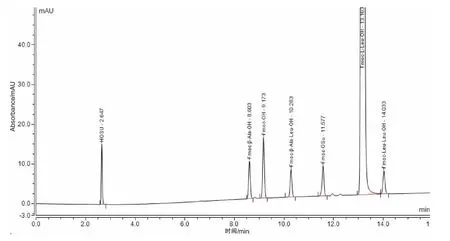
图1 系统适用性溶液色谱图
(2)系统重复性:精密称取Fmoc-L-Leu-OH 对照品适量,加乙腈溶解并定量稀释制成每毫升中约含Fmoc-L-Leu-OH 1.0 微克的对照溶液(0.1%),按“2.2色谱条件”连续进样检测6 次,结果显示峰面积的RSD 为0.69%,说明系统稳定,系统重复性较好。
(3)检测限及定量限:取Fmoc-L-Leu-OH 对照品及各杂质对照品,分别用稀释液逐级稀释至适宜浓度按前文所述“色谱条件”进样检测,分别以信噪比3∶1和10∶1 时注入仪器的量作为检测限和定量限。结果发现,各杂质的检测限均不高于主成分质量的0.0040%,定量限均不高于主成分质量的0.0100%,表明色谱条件灵敏度高,适合进行杂质控制。
(4)定量限重复性:取“(3)检测限及定量限”中规定的Fmoc-L-Leu-OH 对照品稀释至定量限的溶液按前文所述“色谱条件”连续进样6 次,结果主峰保留时间RSD 为0.03%,峰面积的RSD 为5.63%,表明定量限重复性良好。
(5)线性和范围:分别精密量取各定位溶液适量,置于同一个10 mL 量瓶中,定容,摇匀,即得混合对照品储备液。分别精密量取混合对照品贮备液适量,稀释制成每毫升中含Fmoc-L-Leu-OH 和各已知杂质均为0.1、0.5、1.0、1.5、2.0、3.0 微克的溶液,取Fmoc-LLeu-OH 和各已知杂质的定量限溶液和上述系列浓度溶液进样检测,以浓度(微克/毫升)为x轴,峰面积A为y轴,进行线性回归,结果发现Fmoc-L-Leu-OH 和各已知杂质在定量限至3 微克/毫升范围内呈现良好的线性关系。根据标准曲线斜率计算校正因子(校正因子=Fmoc-L-Leu-OH 的标准曲线斜率/杂质的标准曲线斜率),HOSu、Fmoc-OSu、Fmoc-OH、Fmoc-β-Ala-OH、Fmoc-β-Ala-Leu-OH、Fmoc-Leu-Leu-OH的杂质校正因子在0.2~5.0,因此可采用加校正因子的主成分自身对照法计算各杂质的含量。
(6)准确度:已知供试品溶液中Fmoc-OH、Fmoc-Leu-Leu-OH 含量分别约为0.03%、0.06%。取供试品溶液适量,分别加入各已知杂质对照品,并用乙腈稀释至刻度,使各杂质的含量为质控限度的80%、100%、120%,每个浓度各配制3 份,并分别配制自身对照溶液(0.1%),按前文所述“色谱条件”进样检测。计算平均回收率和RSD,结果发现各杂质均能被准确测出。
(7)精密度:
①重复性:精密称取待测样品适量,平行配制6份供试品溶液按前文所述“色谱条件”进样检测,结果显示6 份供试品溶液均仅检测出Fmoc-OH、Fmoc-Leu-Leu-OH 两个杂质,两个杂质的RSD 分别为3.22%、3.58%,表明本方法重复性较好。
②中间精密度:在不同时间,由不同操作人员平行配制6 份供试品溶液,采用不同仪器考察样品的有关物质和杂质个数,与重复性试验的6 份实验结果比较,结果显示6 份供试品溶液检测出Fmoc-OH、Fmoc-Leu-Leu-OH 两个杂质,两个杂质的RSD 分别为3.35%、2.48%,两个操作人员共12 份供试品溶液Fmoc-OH、Fmoc-Leu-Leu-OH 两个杂质的RSD 分别为3.81%、3.00%,表明本方法中间精密度良好。
(8)溶液稳定性:取供试品溶液于室温下放置0、2、4、6、8、10、12 小时,按色谱条件分别进样检测,按面积归一化法计算各杂质的含量,结果显示供试品溶液在室温放置12 小时后,已知杂质Fmoc-OH、Fmoc-Leu-Leu-OH 无增长,其他已知杂质、其他单杂均未检出,溶液稳定,溶剂峰无干扰。
(9)耐用性:在不同流速(0.9、1.0、1.1 毫升/分)、柱温(32、35、38℃)及色谱柱(Waters XSelect CSHTM C18 4.6×250 毫米,5 微米柱、岛津GL WondaSil C18,4.6×250 毫米,5 微米)条件下,系统适用性溶液各峰的相对迁移时间、理论塔板数均无明显变化,各峰分离度均大于2,能满足杂质的分离要求,且供试品溶液有关物质检测结果基本一致。
三、结论与展望
本文对高效液相色谱法测定Fmoc-L-Leu-OH 中的有关物质进行系统的方法学验证,验证结果表明:该方法空白溶剂无干扰,Fmoc-L-Leu-OH 中的6 种已知杂质能达到有效分离;6 种杂质的检测限为主成分质量的0.0006%~0.0040%,定量限为主成分质量的0.0020%~0.0100%,可有效检测出各杂质;各杂质在定量限至3 微克/毫升范围内线性关系良好,线性相关系数r均大于0.999,平均回收率在91.6%~103.6%(n=9),可准确检测各杂质;且精密度与系统重复性良好;该方法对流速、柱温及色谱柱耐受性良好,可用于Fmoc-L-Leu-OH 中有关物质的检测和控制。
由于多肽原料药起始物料的杂质水平直接关系到目标药物的安全性和有效性,在其分析方法开发过程中,应充分考虑药物特点、风险评估、灵敏度目标、可接受误差及试验可行性等,合理选择分析方法和平台,提高分析方法开发的效率,有效地对原料药起始物料进行质量控制。
猜你喜欢
昆钢科技(2021年2期)2021-07-22
艺术品鉴(2020年6期)2020-12-06
职工法律天地(2018年12期)2018-01-22
现代园艺(2017年13期)2018-01-19
水利信息化(2017年4期)2017-09-15
领导文萃(2017年6期)2017-03-24
中学生数理化·高一版(2016年7期)2016-12-07
现代检验医学杂志(2016年3期)2016-11-15
中学生数理化·中考版(2015年12期)2015-09-10
药学与临床研究(2015年4期)2015-06-05
