
微囊藻毒素转运酶MlrD结构及生物学特性分析
2023-09-20 10:08王瑞萍岳士忠于家峰李洁明
生物信息学 2023年3期
王瑞萍,岳士忠,于家峰,李 季,李洁明*
(1.山东省生物物理重点实验室(德州学院 生物物理研究院),山东 德州 253023;2.中国农业大学 资源与环境学院,北京 100193)
微囊藻毒素(Microcystins, MCs)是蓝藻中出现频率最广和危害最大的一种单环七肽肝毒素[1],分子量在1 000 左右,一般结构见图1,其中A、B是可变氨基酸,Adda的分子式为3-氨基-9-甲氧基-2,6,8-三甲基-10-苯基-4,6-二烯酸,是MCs潜在的毒性基团[2]。由于A、B的可变性以及其它氨基酸甲基化与去甲基化的差异,使得MCs具有多种亚型[3-4],到2017年已发现240余种[5-6],其中MC-LR、-RR、-YR(L,R,Y分别代表亮氨酸、精氨酸、酪氨酸)在天然水体中出现频率最高,MC-LR毒性最大,其次为-RR,-YR[7]。MCs主要作用器官是肝脏,对生态系统及人类健康存在极大威胁。生物降解去除MCs因其安全、高效、成本低、无污染等特点引起了国内外的广泛研究。

图1 MCs化学结构式[2]Fig.1 Chemical structure of MCs [2]
MCs的生物降解是在蛋白酶催化作用下完成的,目前的研究主要集中在基因功能方面。早期利用分子生物学手段研究发现,MCs降解菌Sphingomonassp. ACM-3962 (MJ-PV) 在降解MC-LR的过程中,mlr基因簇发挥了重要作用,其中mlrA表达的水解酶(MlrA)通过断裂Adda-Arg键将环状MC-LR水解为线性,随后mlrB表达的酶(MlrB)将线性MC-LR的Ala-Leu键断裂,使其进一步裂解产生四肽,最终四肽在mlrC表达的酶(MlrC)的催化作用下裂解为更小片段[8- 9]。后续通过异源表达的方式在其他降解菌中证实了mlrA、mlrB、mlrC的功能,同时表明MlrC 能够直接降解线性MC-LR和-RR而无需MlrB的作用[10-12]。最初的研究表明,mlrD和mlrA组成一个操纵子,并推论mlrD基因能够编码一种转运蛋白[8],但到目前为止并没有进一步研究mlrD基因的功能。本课题组曾试图通过基因敲除的方式研究野生菌Novosphingobiumsp. THN1中mlrD基因的功能,但因同源双交换难以发生以及敲除后的菌株很快死亡,导致后续功能验证无法继续进行。通过生物信息学的手段探究MlrD结构特征,预测其活性位点,可为进一步研究其功能奠定基础。
对蛋白质序列进行分析有助于了解其结构和功能,进而为研究生物学功能提供关键信息[13]。Bourne等在发现mlr基因簇的同时,对ACM-3962降解酶Mlr蛋白的性质进行了分析[8],表明MlrA包括336个氨基酸残基,分子量为36.6 kD,预测等电点为9.76,存在信号肽,裂解位置在第26和27个氨基酸之间,之后通过序列比对预测并确认了其活性位点H260AIHNE265[14]。Xu等构建了MlrA的三级结构,并通过分子对接和定点突变验证了其他活性位点(Glu172、Trp176、His205、Trp201)[15]。Bourne等[8, 16]对MlrC蛋白序列进行分析,发现并证实了活性位点Asp167、His169、His191。Wang等[17]通过定点突变确定了其他几个活性位点,同时结合MlrC的三级结构进一步分析了活性位点在空间上的分布,推测了其活性口袋。
早期研究表明MlrD包含422个氨基酸残基,分子量为45.4 kD,预测等电点为9.83,有12个跨膜结构域,序列相似性结果显示MlrD属于PTR2超家族[8]。MCs降解酶MlrA、MlrB和MlrC的功能已经被证实,但被认为是转运蛋白的MlrD的功能及结构依然是未知的。近年来,随着基因组学和蛋白质组学的发展,数以万计的蛋白质三维结构尚待分析,但蛋白质结构的确定非常困难[18]。随着计算机技术的发展和已知三维结构蛋白的增多,对未知结构蛋白的预测成为可能。平均50%~70%的典型基因组可以使用计算技术进行结构建模[19]。预测蛋白质结构将有助于探索其活性位点及作用方式,进一步多角度了解MCs的降解。本研究在前人研究的基础上,全面的分析mlrD的序列特征,构建其三级结构,预测活性位点,为进一步研究mlrD的功能及结构提供理论依据。
1 数据来源及研究方法
1.1 MlrD注释及其蛋白序列特征分析
通过在NCBI数据库中检索并下载降解菌的ACM-3962的MlrD(MlrD-ACM-3962)蛋白序列,分别利用在线软件Cell-PLoc 2.0和Protscale分析该蛋白亚细胞定位和亲/疏水性。利用 STRING 数据库对蛋白相互作用关系进行分析。
在NCBI数据库搜索并下载MCs降解菌的MlrD及16S rDNA序列,同时下载外围基因的相应序列。用clusterW[20]进行多序列比对,MEGA7.0构建系统发育树,对mlrD亲缘关系和进化关系进行分析。
1.2 MlrD蛋白三级结构分析
利用HMMTOP工具分析MlrD二级结构,在PDB数据库中检索与MlrD-ACM-3962高度相似的蛋白,筛选模板蛋白并下载其三级结构,选取easymodeller 4.0(Modeller 9.20)[21]软件建模。首先将模板蛋白导入软件,通过模板比对、模板与目的蛋白比对等过程,构建9个模型。构建好的模型用Ramachandran图和Verify-3D[22-23]程序进行评估。评分都通过的模型作为最终的蛋白结构模型,并在PyMOL2中分析展示。
1.3 MlrD蛋白活性位点分析
在上述结构分析的基础上,进一步分析MlrD的保守结构,通过同一蛋白家族已有蛋白活性位点的分析,推测MlrD的活性位点。
2 结果与分析
2.1 MlrD-ACM-3962蛋白基本性质分析
对ACM-3962的MlrD蛋 白 的 一 级 结 构 进 行ProtScale 分析,该多肽链的第 182位 异亮氨酸(Ile)和第361位的半胱氨酸(Cys)分别具有最高分值(1.500)和最低分(-2.867),即前者疏水性最强,后者亲水性最强 (见图2a)。利用 ProtParam 分析可知,氨基酸序列平均疏水指数为0.677,即该蛋白为疏水性蛋白。使用Cell-PLoc 2.0 在线网站分析目标蛋白亚细胞定位,结果表明此蛋白质位于细胞内膜上。

图2 MlrD蛋白亲水性及互作网络分析Fig.2 Analysis of hydrophilicity and interaction network of MlrD protein
蛋白互作网络中的SCI_04307为MlrD蛋白序列,SCI_03700、nadK和SCI_01691与MlrD 相互作用(见图 2b ),其中 nadK 得分最高说明其与 MlrD 蛋白相互作用更紧密。nadK参与调节NAD和NADP的胞内平衡,是NADP生物合成的关键酶,特异性催化NAD腺苷部分2'-羟基的磷酸化生成NADP;SCI_03700是 NADH焦磷酸酶,属于Nudix水解酶家族;SCI_01691为二肽/三肽转运酶。根据互作网络推测,MlrD可能参与了NADH的合成,MCs的转运可能是在MlrD与SCI_01691的协同作用下完成的。
2.2 MlrD的同源性分析
在ACM-3962菌株中首次鉴定出mlrD基因后,后续研究根据已经发现的序列,通过PCR或基因组测序,在其他降解菌中也发现了mlrD基因序列。本文通过在NCBI数据库中检索MCs降解菌的mlrD的基因序列,对降解菌及MlrD蛋白注释进行了总结(见表1)。目前分离到的携带mlrD基因的降解菌共有17株,都属于α变形菌门,其中鞘氨醇盒菌属(Sphingopyxissp.)有9株,占有最高的比例。由于有些菌株只测了mlrD基因的部分序列,所以在序列长度方面差异比较大。对mlrD的基因注释并不统一,可以分为三类,第一类(8株)注释为微囊藻毒素降解酶(包括THN1菌株中较为模糊地注释为参与了微囊藻毒素的降解);第二类注释为肽转运蛋白,其中6株为寡肽转运蛋白,1株(Novosphingobiumsp. MD-1菌株)注释为二肽/三肽转运蛋白,1株(Sphingosinicellasp. strain JEZ-8L菌株)较为确定的注释为微囊藻毒素转运蛋白;第三类未进行注释(菌株Sphingopyxissp. LH21 和Sphingopyxissp. C-1)。
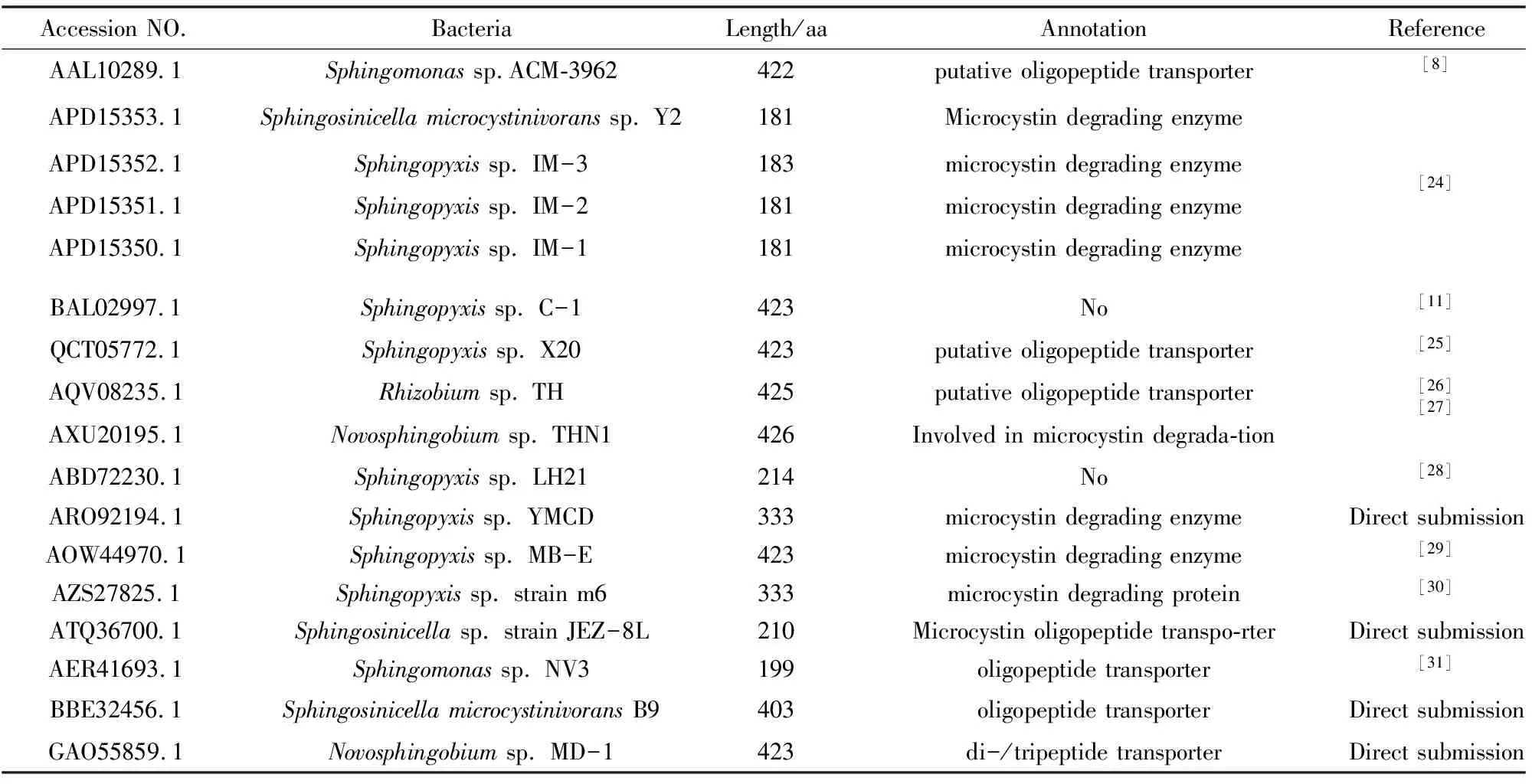
表1 MCs降解菌中MlrD 的特征Table 1 Characteristics of MlrD in diverse MCs-degradation bacteria
为进一步分析不同菌株之间MlrD的保守性,用clustal W进行了多序列比对,结果表明序列较短的MlrD是长序列中的一部分,不同菌株之间的MlrD是保守的。采用MEGA7.0 Neighbor-Joining法对mlrD基因及相应菌株的16S rDNA序列构建系统发育树,选取Myxococcusfulvus124B02和Nitrosomonadalesbacterium0125_3作为外围基因,在mlrD基因发育树中,降解菌形成了三个分支。除Sphingopyxissp.X20以外,Sphingopyxis属的降解菌形成了一个独立的分支(Clade I),Sphingomonas属和Novosphingobium属的降解菌形成了一个分支(Clade II)(见图3a),这两个分支具有较近的亲缘关系,Sphingosinicella属和Rhizobium属形成了另一个分支(Clade III)。16S rDNA基因发育树的结构(见图3b)与mlrD基因相同,也形成了三个独立的分支,除Sphingopyxissp.X20和SphingosinicellamicrocystinivoransB9的分支位置不一致外,其余菌株分支与mlrD基因完全相同。

图3 不同 MCs 降解菌系统发育分析Fig. 3 Phylogenetic analysis of various MCs-degrading bacteria
2.3 MlrD蛋白三级结构分析
在Bourne等[8]对MlrD-ACM-3962初步分析的基础上,构建了其三级结构模型。将MlrD-ACM-3962序列与PDB数据库中蛋白序列进行比较,根据比对的结果,发现与MlrD相似的蛋白较多,相似率在30%左右,大部分属于肽转运蛋白,充分利用较多的相似蛋白,采用Modeller多模板建模预测其三级结构。Modeller同源建模使用多个模板可以组合来自各个模板结构的信息,通常会提高模型的准确性[21]。
根据相似性,以4ikxa、4ikva、4q65a、4oh3a、4xnia、4apsa、5mmta、4lepa、4w6va、6exsa、2xuta、6ei3a、5a2na蛋白为模板,通过模板蛋白互相比对、与目的蛋白比对、主链生成、loop区建模、模型优化[32],用Modeller构建9个模型。9个模型分别用Ramachandran 图和Verify-3D评分进行评价,Ranmachandran 图在最允许范围内氨基酸所占比例达到90%以上,被认为是一个高质量的模型[33],Verify-3D 分析原子模型与自身序列在位置和环境上的兼容性,如果 80%的氨基酸残基得分大于等于0.2,可认为模型是合理的[34]。经过比较,选取评分较高的模型(见图4a),从Ramachandran图统计结果(见图4b)可以看出,模型中93.0%的氨基酸在最允许区域内,5.9%在额外允许区域。Verify-3D评估结果表明85.78%的残基3D-1D评分大于等于0.2(见图4c),即相容性是通过的。综上MlrD的三维结构具有一定的理论意义和参考价值。
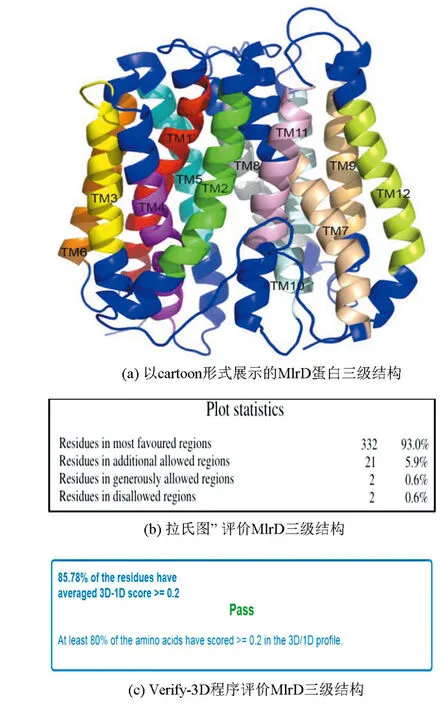
图4 MlrD蛋白三级结构 及其评价Fig.4 The 3D structure of MlrD and its evaluation
MlrD由422个氨基酸组成,HMMTOP二级结构预测表明由12个跨膜α-螺旋和无规则卷曲组成,没有β-折叠结构。在12个跨膜α-螺旋中,前6个与后6个跨膜α-螺旋结构域呈现出非常相似的拓扑结构,以一种假二次轴对称的方式存在,对称轴垂直于膜平面
2.4 MlrD蛋白功能及活性位点分析
经过保守结构域的查找,发现MlrD蛋白属于质子依赖型寡肽转运蛋白PTR2(Peptide transport)超家族,这个家族的蛋白属于二肽/三肽渗透酶,参与氨基酸运输与代谢。
PTR2家族有两个保守基序(Motif)序列,分别是位于第2和3跨膜螺旋连接处的GXXXADXXXGKXXTI(X代表任意氨基酸)和位于第5个跨膜螺旋上的FYXXINXG(X代表任意氨基酸),第二个保守motif在肽转运家族的所有成员中都表现出绝对的保守性[35-36]。在MlrD序列中这两个motif与PTR2具有一些差异 (见图5),第1个保守序列位于第2和3跨膜螺旋连接处,位于MlrD-ACM-3962的69-83 bp,氨基酸序列为GGWIADRFIGRSAAI,即上述保守序列中的K和T分别变成了R和A;第2个保守序列在MlrD中不存在,比较相似的序列位于第5个跨膜螺旋上142 -150 bp,氨基酸序列为FYYLAVSAG。这两个序列中个别氨基酸的差异可能是由于底物的差异性造成的,这些序列在不同MCs降解菌之间是保守的(见图5)。综上所述,位于第2和3跨膜螺旋连接处的Gly69、Ala73、Asp74、Gly78、Arg79、 Ala82、Ile83和位于第五个跨膜螺旋上的Phe142、Tyr143、Ala146、Val147、Gly150可能为MlrD的活性位点。
3 讨 论
到目前为止,MlrD的功能并未通过实验证实,对其基因注释大多是根据序列保守性分析或依据前人的经验得到的,导致注释存在差异。
系统发育树分析表明mlrD和16S rDNA具有相同的拓扑结构。Qin等[25]的研究表明其他mlr基因构建的系统发育树都形成了三个主要分支,而且整体结构相同,表明mlrD基因与其他mlr基因具有相同的进化来源,整个mlr基因簇共同进行进化。 经过多序列比对及系统发育树分析,表明MlrD在MCs降解菌之间是高度保守的,MlrD 具有功能特异性。
Dziga等通过异源表达的方式研究MlrA、MlrB、MlrC的功能[10, 11, 14, 16-17, 37],表明在没有mlrD基因的情况下,菌株能够降解MCs及其产物,说明在异源表达菌株中mlrD基因对于MCs及其产物的转运和降解不是必需的,然而在异源表达菌株中的情况并不适应于野生菌。利用同源双交换敲除基因是研究野生菌基因功能的常用方法之一,但在对mlrD进行敲除时,因二次同源双交换难以发生,双交换菌株不稳定等原因,基因敲除没有成功,表明MlrD是菌株生长所必需的,大片段的缺失会导致菌株的死亡。通过生物信息学的方法预测MlrD活性位点,进一步采用活性位点碱基突变、敲除等方法,可在野生菌中进一步研究MlrD的功能。
保守结构域分析表明MlrD属于PTR2超家族,在膜转运蛋白分类数据库(TCDB)中,PTR是位于主要协助转运蛋白超家族(MFS)内的一个蛋白家族。MFS家族的蛋白大多数都含有400-600个氨基酸残基,大多数拥有12个跨膜α-螺旋[38],N端和C端分别由6个跨膜α螺旋组成,呈现出非常相似的拓扑结构,以假二次轴对称的方式存在[39-40]。MlrD的结构完全符合MFS家族的特性。MlrD的结构与上述特征相符合,从三级结构的角度说明MlrD属于MFS超家族。
MFS家族的蛋白都是次级转运蛋白的典型代表,利用膜内外底物浓度差产生的电化学势能转运底物[41-42], MCs及其产物可能通过浓度差在MlrD的作用下进出细胞。有研究表明,突变MFS上述两个保守序列会导致转运活性丧失[43-44],因此后续研究可以通过定点突变的或小片段基因敲除方法,突变或敲除GGWIADRFIGRSAAI和FYYLAVSAG这两个区域,深度了解MCs及其降解产物的转运。
4 结 论
MCs降解菌的mlrD和16S rDNA系统发育树有相同的拓扑结构,MlrD蛋白属于PTR2超家族,其二级结构由α-螺旋和无规则卷曲结构组成,且完全符合PTR2家族的特性,可能的活性位点为位于第2和3跨膜螺旋连接处的Gly69、Ala73、Asp74、Gly78、Arg79、Ala82、Ile83和位于第5个跨膜螺旋上的Phe142、Tyr143、Ala146、Val147、Gly150,可以通过突变或敲除这两个结构域来研究其功能。
猜你喜欢
上海金属(2021年6期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
医药前沿(2020年28期)2020-12-02
中国洗涤用品工业(2019年4期)2019-05-11
心肺血管病杂志(2019年1期)2019-04-22
生物学通报(2019年3期)2019-02-17
中成药(2018年1期)2018-02-02
教学考试(高考生物)(2017年4期)2017-12-13
动物医学进展(2015年10期)2015-12-07
食品工业科技(2014年7期)2014-03-11
