
等离子体辅助铈基催化剂催化氨制氢的性能研究
2023-09-19 05:14高一博胡二江殷阁媛黄佐华
石油学报(石油加工) 2023年5期
高一博,胡二江,殷阁媛,黄佐华
(西安交通大学 动力工程多相流国家重点实验室,陕西 西安 710049)
氢(H2)被普遍认为是最有前途的能源载体之一,被广泛应用于各种工业领域[1-2]。H2具有高度易燃、易爆和低压缩的特点,在储运方面存在诸多困难,极大限制了其规模化应用[3-4]。氨(NH3)具有较高的体积能量密度和质量能量密度,被认为是一种可行的储氢介质[5-6]:①NH3作为一种零碳燃料(2NH3→3H2+N2,ΔH=92.8 kJ/mol),符合“碳达峰,碳中和”的战略目标;②NH3具有高储氢量(氢质量分数高达17.8%)、高体积密度(121 kg/m3的液体形式)、高能量密度(约3 kWh/kg)和丰富的产量;③NH3液化发生在较温和的条件下,所以NH3的储运较易操作。因此,氨分解制氢研究受到了越来越多的关注。
目前,广泛采用的氨分解方法主要集中在热催化法[7-8]、电解法[9-10]、光解法[11-12]和等离子体法[13-14]等。以氨分解制氢为目的的研究主要是通过热催化法。热催化氨分解性能最优的催化剂是Ru基金属催化剂[15],因为Ru具有最佳的金属-氮(M-N)的键能强度,有利于NH3分解和N2脱附。但Ru基金属的稀缺性和高成本限制了其大规模应用,需要大力开发可替代的廉价金属催化剂[16-17]。3d金属Fe基[18-19]、Co基[20-21]和Ni基[22-23]催化剂已被证实可作为Ru基替代金属催化剂高效催化氨分解制氢。但是,非贵金属基催化剂催化氨分解的反应活性通常比Ru基催化剂低得多。因为Co-N和Ni-N的M-N结合能低于Ru-N,而Fe-N和Mo-N的结合能高于Ru-N[15]。因此,设计制备3d双金属催化剂(M-N结合能为597.1 kJ/mol)是提高氨分解制氢效率的有效方法。
作为一种常见的稀土金属氧化物,CeO2具有优良的储放氧能力和金属阳离子转化效率(Ce3+↔ Ce4+),通常作为催化剂,载体和启动子用于化学反应(如碳烟脱除、合成氨和甲醇重整制氢等)[24-25]。Hu等[26]研究CeO2载体可以有效吸附NH3;Muroyama等[27]报道了Ni负载的CeO2基催化剂可以高效催化NH3的分解转化;而Zhou等[28]报道了Ni/CeO2-BN催化剂中BN的修饰可以促进NH3分解。虽然CeO2基催化剂已被报道用于催化NH3分解产氢,但研究尚少,同时分解温度较高且选择性较低。因此,需要开展廉价3d过渡金属掺杂CeO2基催化剂低温氨分解性能研究。
除催化剂外,在介质阻挡放电(Dielectric barrier discharge,DBD)反应器中,非热平衡等离子体(Non-thermal plasma,NTP)同样能够降低氨的分解温度,促进氨的分解和转化[8,14]。NTP不仅增强了活性金属对NH3的吸附,也加速了表面吸附N原子的复合解吸。
1 实验部分
1.1 原料和试剂
六水合硝酸铈(Ⅲ)(Ce(NO3)3·6H2O,纯度99%)、九水合硝酸铁(Ⅲ)(Fe(NO3)3·9H2O,纯度98.5%)、六水合硝酸钴(Ⅲ)(Co(NO3)·6H2O,纯度99%)、六水合氯化镍(Ⅱ)(NiCl2·6H2O,纯度98%)和氢氧化钠(NaOH,纯度95%),购自阿拉丁化学有限公司。乙二醇、聚乙烯吡咯烷酮(PVP)和碳酸钠(Na2CO3·H2O),购自麦克林化工有限公司。
1.2 催化剂制备
参照文献[29]的方法制备CeO2纳米球(CeO2-S):将含有Ce(NO3)3·6H2O(1.00 g)的蒸馏水(2 mL)溶解在聚乙烯吡咯烷酮(0.40 g)和乙二醇(30 mL)的混合溶液中搅拌0.5 h,将溶液倒入体积为50 mL的高压反应釜中,160 ℃加热8 h,反应釜冷却至25 ℃后,收集所得淡黄色粉末,用蒸馏水离心洗涤3次。最后,于60 ℃真空干燥,干燥后的淡黄色粉末即为CeO2-S样品。
参照文献[30]采用沉积-沉淀法制备Fe/CeO2-S样品:首先在蒸馏水(15 mL)中加入CeO2-S(1.00 g)和Fe(NO3)3·9H2O(1.45 g)(Fe负载质量分数5%),加入碳酸钠(0.5 mol/L),将pH值调至9。收集沉积物,用蒸馏水离心洗涤3次,在60 ℃下真空干燥后,在马弗炉中450 ℃煅烧4 h,即得到Fe/CeO2-S样品。
Co/CeO2-S和Ni/CeO2-S的金属前驱体分别由Co(NO3)2·6H2O和NiCl2·6H2O取代。采用与Fe/CeO2-S相同的制备方法,获得3d过渡金属Fe、Co和Ni分别共掺杂CeO2-S催化剂FeCo/CeO2-S、FeNi/CeO2-S和CoNi/CeO2-S。催化剂中Fe/Co、Fe/Ni、Co/Ni摩尔比均为1。
1.3 催化剂表征
采用德国Bruker D8 Advance型X射线衍射仪(XRD)测定催化剂的晶体结构,CuKα辐射(λ=0.15418 nm)。扫描速率为6 °/min,每隔0.02°记录20°~90°的2θ数据。
采用美国Micromeritics ASAP 2020 Plus HD88型比表面积分析仪进行N2吸附-脱附测试。通过传统的Brunauer-Emmett-Teller(BET)和Barrett-Joyner-Halenda(BJH)模型得到催化剂颗粒的比表面积(SBET)、孔体积(Vp)和平均孔径(Dp)。
采用美国Thermo Scientific Talos-F200X型透射电镜,获得催化剂的高分辨率透射电镜(HRTEM)、高角度环形暗场扫描透射电镜(HAADF-STEM)照片和能量色散X射线能谱(EDX-mapping)。
采用美国Thermo Scientific K-Alpha型X射线光电子能谱仪(XPS)获得催化剂的XPS谱图,单色AlKα(1486.7 eV)作为X射线源。
采用美国Micromeritic AutoChem1Ⅱ2920型化学吸附仪进行H2-TPR测试。50 mg催化剂在He流量50 mL/min下500 ℃预处理0.5 h;预处理后,催化剂在100~900 ℃范围内以升温速率10 ℃/min升温,在5% H2/He流量为50 mL/min下还原。
采用美国Thermo Scientific Nicolet iS50型红外光谱仪(FT-IR)和汞镉碲化探测器,在配有CaF2窗口的反应池(Harrick)中进行FT-IR分析。以NH3为探针,在300 ℃下进行稳态模式的FT-IR测试;在测试之前,新鲜样品在300 ℃下经过5% H2/Ar处理0.5 h;首先对催化剂进行预处理使其处于稳态模式,气体混合物(10%NH3/Ar)以体积流量10 mL/min 在300 ℃下通过催化剂20 min。
采用美国Thermo Fisher Scientific公司生产的DXRxi拉曼光谱仪进行拉曼光谱分析,仪器采用超低暗噪声和单光子信号水平检测的EMCCD检测器。
1.4 氨分解催化性能评价实验
催化剂和等离子体催化氨分解实验装置示意图如图1所示。采用壁厚为2 mm、外径为19 mm的石英管作为介质层,外电极采用接地铜环包裹在石英管外表面。将直径为5 mm的铜棒固定在管轴上,作为内电极连接高压纳秒脉冲电源。该系统的放电长度为30 mm。为了减少催化剂对内电极的影响,在内电极周围包裹了一根1 mm厚的石英管。内部电极连接高压和电流探头,用于测量放电过程中的电压和电流数据。电极上产生的2个电信号都显示于数字示波器上。

MFC—Mass flowmeter control;GC—Gas chromatography;PC—Personal computer;FT-IR—Fourier transform infrared spectoscopy图1 介质阻挡放电等离子体催化氨分解实验装置示意图Fig.1 Schematic diagram of experimental setup for ammonia decomposition catalyzed by dielectric barrier discharge plasma
采用固定床反应器(长450 mm,内径15 mm)进行CeO2基催化剂(用量0.5 g,40~80目)的介质阻挡放电等离子体氨分解催化性能评价实验。实验前,催化剂在Ar气(体积流量100 mL/min)下500 ℃预处理1 h。冷却至25 ℃后,加入5%NH3/Ar(体积流量300 mL/min),反应气体体积空速(GHSV)为36000 cm3/(gcat·h)。反应区的温度区间为50~700 ℃。在无催化剂的情况下进行空白实验。采用美国Bruker INVENIO-R型FT-IR仪实时监测和记录NH3含量。采用美国Agilent 7890B型气相色谱仪(GC)和热导率检测器(TCD)在线检测排放气体。为测试催化剂的热稳定性,将催化剂加热温度保持在600 ℃测试48 h。
NH3的转化率(xNH3,%)定义如式(1)所示。产氢速率(RH2,mmol/(gcat·min))定义为每克催化剂每分钟产生氢气的物质的量,如式(2)所示。
(1)
(2)
式中:nNH3为转化NH3的物质的量,mmol;nNH3,t为总NH3的物质的量,mmol,FNH3为NH3的体积流量,mL/min;mcat为催化剂质量,g;Vm为标准摩尔体积(22.4 L/mol)。
催化剂各活性位点的转换频率(Turnover frequency,TOF)的计算式如式(3)所示。
(3)
式(3)中:xNH3为450 ℃时NH3的转化率,%;Cas为每克催化剂活性位点的物质的量,mmol/g。
2 结果与讨论
2.1 催化剂表征结果
2.1.1 XRD表征
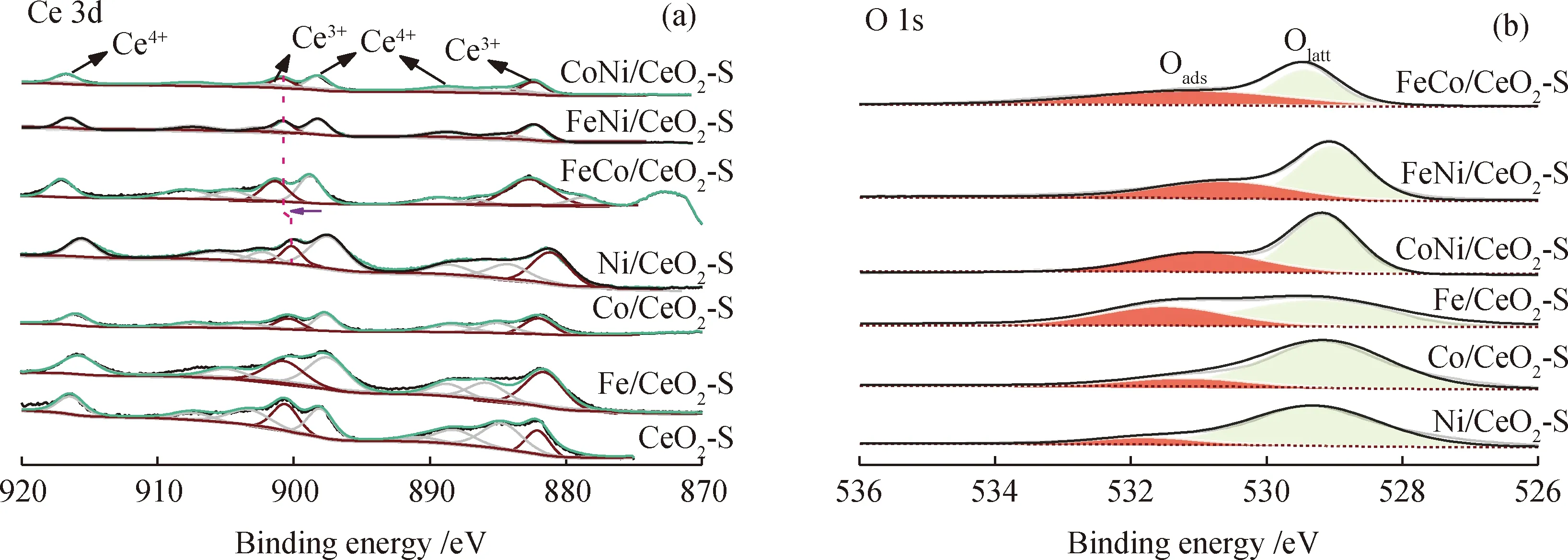
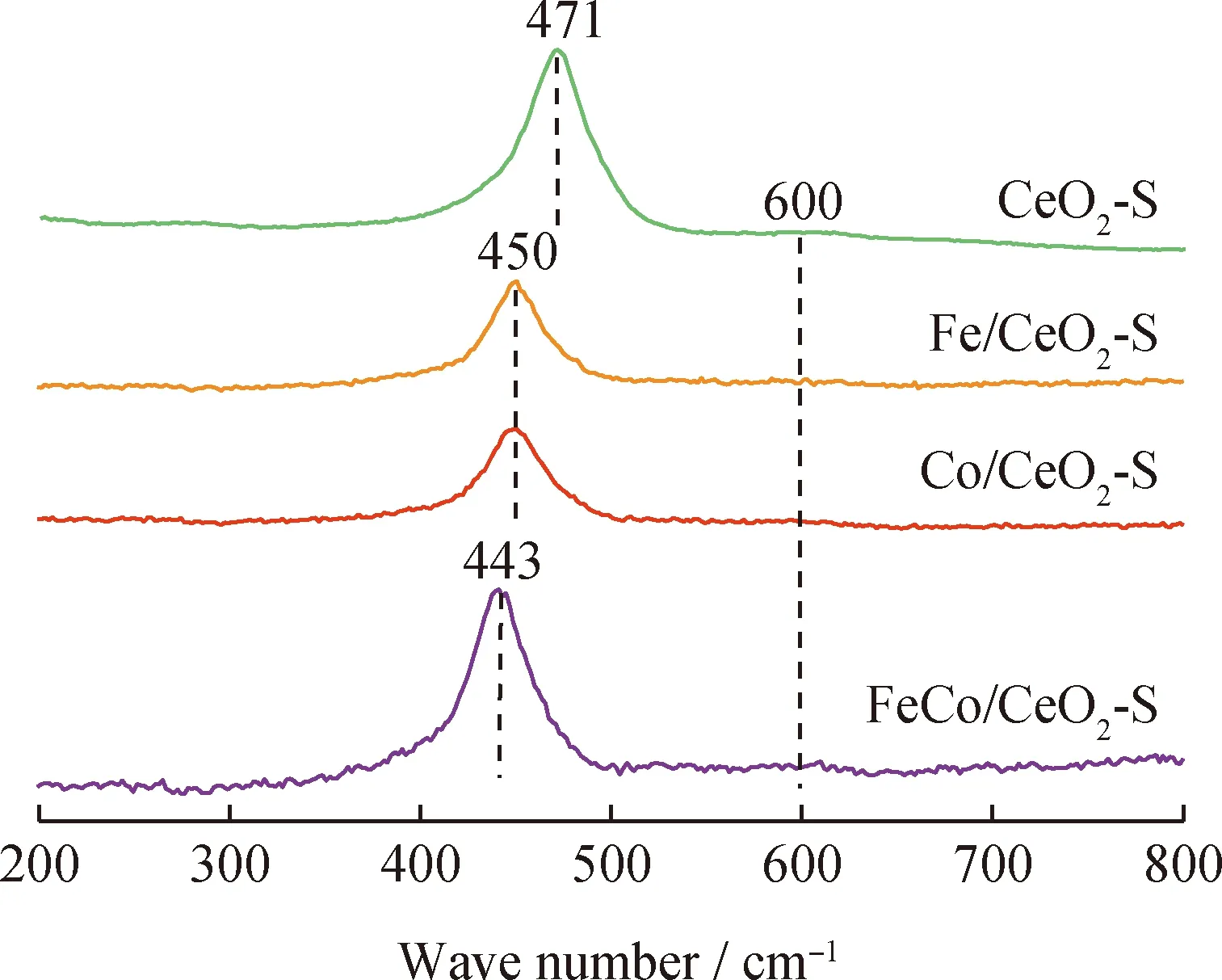
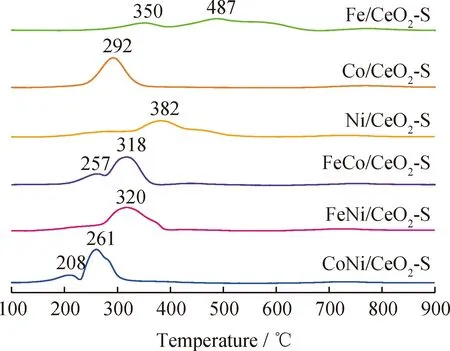
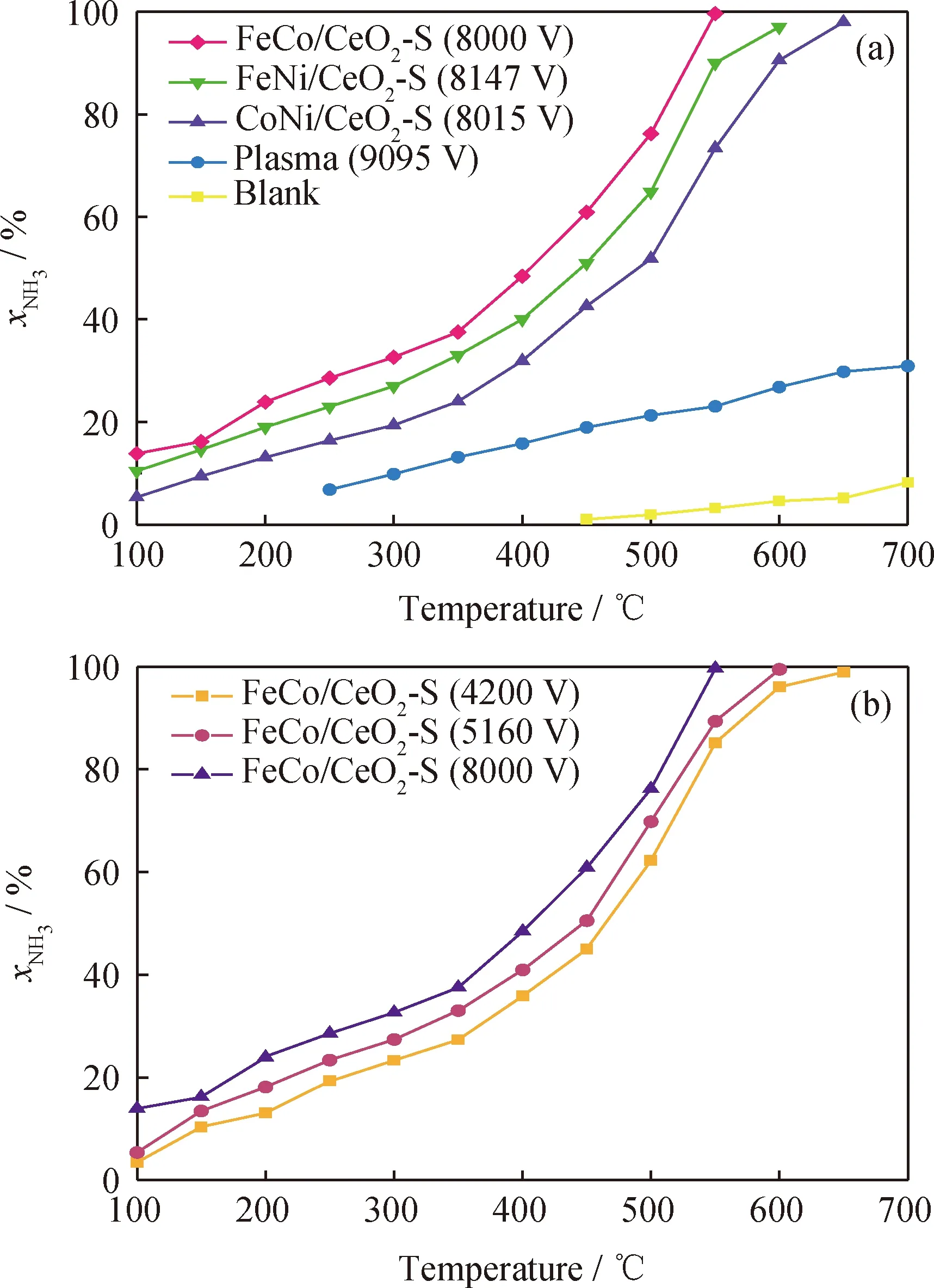
CeO2-S载体和3d过渡金属(Fe、Co、Ni)掺杂CeO2基催化剂的晶体结构如图2所示。CeO2基催化剂的结构参数如表1所示。由图2可见,CeO2基催化剂位于28.6°、33.2°、47.5°、56.3°、59.1°、69.5°、76.9°、79.3°的特征峰分别对应(111)、(200)、(220)、(311)、(222)、(400)、(331)和(420)晶面。根据Debye-Scherrer方程,CeO2-S载体的晶粒直径为19.7 nm。3d过渡金属(Fe、Co和Ni)掺杂CeO2-S载体后,特征峰没有明显的变化,说明3 d金属掺杂到CeO2晶格中形成了稳定的铈基固溶体。同时,Fe/CeO2-S、Co/CeO2-S和Ni/CeO2-S中出现Fe2O3(JCPDS No. 79-0007)、Co3O4(JCPDS No.78-1969)和NiO(JCPDS No.71-1179)的弱特征峰,表明Fe2O3、Co3O4和NiO是煅烧后的主要晶态。单金属掺杂CeO2-S催化剂的晶粒直径由小到大的顺序为:Fe/CeO2-S(4.1 nm) 表1 CeO2基催化剂的结构参数Table 1 Structural parameters of CeO2-based catalysts 图2 CeO2基催化剂的XRD谱图Fig.2 XRD pattern of CeO2-based catalysts 2.1.2 BET表征 图3(a)显示了CeO2-S载体和3d过渡金属(Fe、Co和Ni)掺杂CeO2-S催化剂的N2吸附-脱附等温线。由图3(a)可以看出,所有样品均表现为Ⅳ型等温线和H2型迟滞回线,以Ⅳ型等温线的迟滞回线为介孔结构的标志。对应催化剂BJH孔直径分布如图3(b)所示。由图3(b)可以看出,CeO2-S的孔径分布均匀,平均孔径约为20 nm。在CeO2-S载体上掺杂3d过渡金属(Fe、Co和Ni)后,除FeNi/CeO2-S外,其余孔径均明显增大。 图3 CeO2基催化剂的N2吸附-脱附和孔径(D)分布曲线Fig.3 N2 adsorption-desorption and pore diameter (D)distribution curves of CeO2-based catalysts(a)N2 adsorption-desorption isotherms;(b)Pore diameter (D)distribution 由表1可见:CeO2-S的BET表面积较大,为135.8 m2/g,孔体积为0.382 cm3/g。作为催化剂载体,较大的BET表面积和孔隙结构有利于金属物种的分散,促进氨的分解和活化。FeCo/CeO2-S的BET表面积最大,FeNi/CeO2-S和CoNi/CeO2-S的BET表面积较小。 2.1.3 HRTEM、SAED和EDX-mapping表征 通过HRTEM和SAED得到了CeO2纳米球(CeO2-S)的形貌照片,如图4所示。CeO2拥有3个低指数晶面(100)、(110)和(111)[31-33]。由图4(a)可见,CeO2-S载体呈现出均匀的纳米球结构,形状尺寸为10~55 nm。由图4(b)可见,晶面间距为0.27和0.31 nm,对应于(100)和(111)晶面[34-35]。3d过渡金属(Fe、Co和Ni)共掺杂CeO2-S样品在金属负载和煅烧后均保持了纳米球的形貌。在图4(c)中,SAED显示出Debye-Scherrer环,这归因于立方CeO2样品的反射。反射晶面与XRD表征(Fm-3m JCPDS No.34-0394)的d-spacing值吻合良好。从XRD和SAED分析[35-36]可知,0.31、0.27、0.19、0.16、0.13和0.12 nm的d-spacing值分别对应立方CeO2的(111)、(100)、(110)、(311)、(222)和(400)晶面。通过HAADF-STEM和EDX-mapping 对FeCo/CeO2-S催化剂的元素组成进行了表征,如图5所示。由3d过渡金属(Fe和Co)的元素图像(见图5(d)和(e))可以看出,过渡金属负载元素均匀分布在CeO2纳米球的表面。 图4 CeO2-S催化剂的形貌照片Fig.4 Morphology of CeO2-S catalyst(a)TEM;(b)HRTEM;(c)SAED;(d)Schematic image 图5 FeCo/CeO2-S催化剂的能谱图Fig.5 Energy spectrum of FeCo/CeO2-S catalyst(a)HAADF-STEM;(b)Ce;(c)O;(d)Fe;(e)Co 2.1.4 XPS表征 采用XPS分析了催化剂的阳离子分布状态。Ce 3d 和O 1s的XPS谱图如图6所示。对于Ce 3d光谱(见图6(a)),899.4~901.5 eV和882.0~882.7 eV的峰归属于Ce3+,其余6个峰归属于Ce4+[37-38]。Ce3+的存在是由于电子从CeO2载体转移到掺杂金属(Fe、Co和Ni),Ce3+也被广泛报道用于提高催化活性[39-40]。为了保持电荷守恒,Ce3+的生成通常伴随着氧空位的产生(4Ce4++O2-→4Ce4++0.5O2+2e-/□→2Ce3++2Ce4++0.5O2+□,其中□代表氧空位)。表2为Ce3+、Oads和氧空位(VO)的占比及氧空位特征峰强度(A600/A457)。双金属(FeCo、FeNi和CoNi)共掺杂CeO2-S后,由于更多的电子从载体转移到双金属活性位点,使得Ce3+和氧空位进一步增加。富电子金属物种促进NH3的活化,以及表面N原子的重组实现N2的解吸,从而促进氨分解制氢[41]。 表2 Ce3+、Oads和氧空位(VO)的占比及氧空位特征峰强度(A600/A457)Table 2 Relative area ratio of Ce3+,Oads and oxygen vacancy (VO)and characteristic peak intensity of oxygen vacancy (A600/A457) 图6 3d过渡金属掺杂CeO2-S基催化剂的XPS谱图Fig.6 XPS patterns of CeO2-S-based catalysts doped with 3d transition metals(a)Ce 3d;(b)O 1s 2.1.5 Raman表征 通过Raman光谱进一步测试了CeO2基催化剂的结构特征,Raman光谱谱图如图7所示。对于CeO2-S载体,471和600 cm-1处的散射峰属于铈基氟石型结构的F2g振动模式和缺陷诱导的D-mode峰[28,42]。M/CeO2-S (M为Fe、Co和FeCo)在450和443 cm-1处的红移归因于Fe系金属掺杂到CeO2晶格后引起的晶格收缩。显然,FeCo共掺杂后红移现象增强,表明FeCo与CeO2-S之间的金属和载体之间的强相互作用(Strong metal support interaction,SMSI)增强。如表2所示,M/CeO2-S中氧空位(VO)占比也与缺陷诱导的D-mode峰(600 cm-1)和F2g振动峰(457 cm-1)[43-45]的强度(A600/A457)成正比。结果表明,双金属共掺杂CeO2-S催化剂比单金属共掺杂催化剂具有更高的A600/A457。随着其他3d过渡金属的加入,CeO2中A600/A457增加。FeCo/CeO2-S的A600/A457(0.10)高于Fe/CeO2-S(0.03)和Co/CeO2-S(0.06)。 图7 CeO2-S基催化剂的Raman光谱谱图Fig.7 Raman spectra of CeO2-S-based catalysts 2.1.6 H2-TPR表征 CeO2载体和CeO2基催化剂的还原性通过H2-TPR实验进行研究,结果如图8所示。由图8可以看出,CeO2载体在460~800 ℃观察到2个峰值,这归因于吸附氧和晶格氧的减少[44]。作为单金属掺杂的CeO2-S催化剂,Fe/CeO2、Co/CeO2和Ni/CeO2的还原峰向低温方向移动。Fe/CeO2有2个明显的峰,低温峰(350 ℃)为Fe2O3→Fe3O4的还原,高温峰(487 ℃)为Fe3O4→Fe0还原[45]。Co-O-Ce交联后,292 ℃的还原峰归属于Co3O4→CoO[24]。在382 ℃时,Ni/CeO2-S中只能观察到1个峰。基于SMSI效应[46],Fe2O3、Co3O4和NiO在CeO2-S上的还原一般发生在200~400 ℃。对于双金属共掺杂样品,由于SMSI的存在,其还原峰进一步向低温转移。对于双金属共掺杂催化剂,总耗氢量由大到小依次为:FeCo/CeO2-S(5.33 mmol/gcat)>FeNi/CeO2-S(3.67 mmol/gcat)>CoNi/CeO2-S(3.48 mmol/gcat)。FeCo/CeO2-S表现出最大的H2消耗,表明其优异的氧化还原性能。 图8 CeO2-S基催化剂的H2-TPR曲线Fig.8 H2-TPR profiles of CeO2-S-based catalysts 2.2.1 催化剂催化氨分解 图9(a)为3d过渡金属(Fe、Co和Ni)掺杂CeO2-S催化剂的催化氨分解活性。由图9(a)可以看出,催化性能依赖于掺杂的3d过渡金属(Fe、Co和Ni)。对于单金属掺杂的CeO2-S催化剂,Fe/CeO2-S的催化活性最低。与Co和Ni单掺杂催化剂相比,Fe/CeO2-S的比表面积最小(98.4 m2/g),这与Fe的N解吸势能(619.23 kJ/mol)较高有关。特别对于FeCo/CeO2-S而言,氨在650 ℃时几乎达到完全转化。FeCo/CeO2-S的最佳反应活性不仅因为其最大的比表面积(128.5 m2/g)、孔体积(0.370 cm3/g)和孔径(12.9 nm),还在于富电子的FeCo物种促进了NH3的活化和表面N原子的重组[47]。通过计算转换频率(TOF)以了解CeO2基催化剂的内在活性,结果见图9(b)。由图9(b)可以看出,双金属共掺杂CeO2基催化剂的TOF高于单金属掺杂催化剂。单金属掺杂CeO2基催化剂的氨分解TOF由大到小顺序为:Ni/CeO2-S(0.105 s-1)>Co/CeO2-S(0.063 s-1)>Fe/CeO2-S(0.030 s-1),证明Fe和Co金属在催化氨分解中的固有性质弱于Ni。FeCo/CeO2-S的TOF(0.275 s-1)高于CoNi/CeO2-S(0.205 s-1)和FeNi/CeO2-S(0.246 s-1)。FeCo/CeO2-S具有丰富的氧空位,且Fe-N原子之间的结合能(619.2 kJ/mol)和Co-N原子之间的结合能(523 kJ/mol)适中,因而具有优异的催化活性。 图9 氨分解活性指标曲线Fig.9 Index curve of ammonia decomposition activity(a)NH3 conversion (xNH3)over CeO2-S based catalysts;(b)Turn over frequency (TOF)of CeO2-S based catalysts at 450 ℃,(P+C)represents the synergy of plasma and catalyst;(c)Stability over CeO2-S catalysts at 600 ℃Reaction conditions:5% NH3/Ar flow rate 300 mL/min;m(Catalyst)=0.5 g;GHSV=36000 cm3/(gcat·h) 2.2.2 等离子体与催化剂协同催化氨分解 图10(a)为等离子体辅助氨分解活性指标曲线。由图10(a)可见,与空白对照相比,等离子体降低了氨分解反应温度,而等离子体辅助CeO2-S基催化剂催化氨分解进一步降低了分解温度。特别是对于性能最佳的FeCo/CeO2-S催化剂,由于NH3转化与电子平均能量密切相关,其催化活性随着输入电压的增大而增加[48]。在催化剂和催化剂+等离子体2种模式下,FeCo/CeO2-S催化剂的NH3转化率均高于FeNi/CeO2-S和CoNi/CeO2-S催化剂,其在550 ℃实现了NH3的完全转化,产氢速率RH2为29.3 mmol/(gcat·min)。催化剂+等离子体模式下氨的转化率远高于催化剂和等离子体的单独催化转化,进一步说明介质阻挡放电等离子体的协同促进作用。等离子体辅助M/CeO2-S(M=FeCo、FeNi、CoNi)催化剂催化氨气分解的TOF见图9(b)。等离子体+双金属共掺杂CeO2基催化剂催化氨分解的TOF值由大到小顺序为:FeCo/CeO2-S(P+C)(0.606 s-1)>FeNi/CeO2-S(P+C)(0.512 s-1)>CoNi/CeO2-S(P+C)(0.431 s-1)。 图10 等离子体辅助氨分解活性指标曲线Fig.10 Index curve of plasma-assisted ammonia decomposition activity(a)M/CeO2-S (M=FeCo,FeNi and CoNi)catalysts at different discharge voltages and blank test at discharge voltage of 9095 V;(b)FeCo/CeO2-S catalyst at various discharge voltagesReaction conditions:5% NH3/Ar flow rate 300 mL/min;m(Catalyst)=0.5 g;GHSV=36000 cm3/(gcat·h) (1)合成了3d过渡金属(Fe、Co和Ni)掺杂的纳米球型CeO2基催化剂,并用于NH3分解。随着SMSI的增强,氧空位和Ce3+富集,电子从CeO2载体转移到活性金属位点,加速了N原子的重组。 (2)与单金属掺杂相比,双金属共掺杂CeO2-S催化剂具有更好的氨分解制氢性能。活性最高的FeCo/CeO2-S催化剂在450 ℃时具有最高的转化频率,其最佳活性归因于富氧空位、Ce3+以及Fe与Co之间适中的M-N结合能。在等离子体辅助作用下,FeCo/CeO2-S催化剂在550 ℃实现氨气的完全转化。 (3)催化剂与等离子体的协同效应还取决于输入电压和反应温度。双金属共掺杂FeCo/CeO2-S催化剂形成了中等强度的M—N键和高比表面积,提高了催化活性,达到最高产氢速率(RH2=29.3 mmol/(gcat·min))。





2.2 CeO2基催化剂氨分解催化活性评价结果

3 结 论
猜你喜欢
航天工业管理(2020年9期)2020-12-28
重型机械(2020年2期)2020-07-24
上海建材(2020年12期)2020-04-13
陶瓷学报(2019年5期)2019-01-12
石油化工建设(2018年2期)2018-07-11
当代化工研究(2016年5期)2016-03-20
中国石油大学学报(自然科学版)(2015年2期)2015-11-10
电源技术(2015年11期)2015-08-22
读者欣赏(2014年6期)2014-07-03
语文知识(2014年2期)2014-02-28
