
附子总生物碱纯化工艺优化
2023-09-19 02:44蔡淑慧丁梦磊甘逸夫王洪兰狄留庆
中成药 2023年9期
蔡淑慧,丁梦磊,甘逸夫,王洪兰,张 雯*,狄留庆*
(1.南京中医药大学药学院,江苏 南京 210023;2.江苏省中药高效给药系统工程技术研究中心,江苏 南京 210023)
附子是毛茛科植物乌头AconitumcarmichaeliiDebx.的子根加工品,有回阳救逆、补火助阳、散寒止痛的功效[1],被称为“回阳救逆第一品药”,又被誉为“百药之长”。现代研究表明,附子主要含有生物碱与多糖类成分,具有强心、抗肿瘤等作用[2-3]。生物碱是附子中研究最多、最具疗效的化学成分,该类化合物具有典型的“效-毒” 辨证关系,其含量高低会显著影响附子药效与毒性[4-5]。因此,采用适宜纯化技术获取附子生物碱类成分,对其开发利用具有重要意义。
目前,大孔吸附树脂法是最常用的生物碱纯化手段[6-7],具有选择性好、吸附速度快、机械强度高等优点[8-9]。然而,该方法也受到诸多因素的影响,如树脂类型与上样方案等。因此,优化纯化参数有利于高效节能地获取附子生物碱类成分。星点设计-效应面法[10-11]具有实验次数少、准确度高、预测性好的优势,广泛应用于中药提取与纯化。然而,目前星点设计-效应面法尚未应用于附子生物碱类成分纯化工艺。本实验拟采用单因素考察及星点设计-效应面法优选大孔树脂纯化附子总生物碱的工艺,以期为附子的物质基础研究提供科学依据。
1 材料
1.1 仪器 TU-1810PC 型紫外分光光度计(北京普析通用仪器有限责任公司);PHS-3C 型PH 计(上海仪电科学仪器股份有限公司);Waters 2695 型高效液相色谱仪(美国Waters 公司);CL21R 型微量离心机(美国赛默飞世尔公司);XP-6 型电子天平(百万分之一,瑞士梅特勒-托利多公司);PA1204B 型电子天平(万分之一,上海精密科学仪器有限公司)。
1.2 试剂与药物 黑顺片(产地四川绵阳,批号190715)购自亳州市永刚饮片厂有限公司,经南京中医药大学中药鉴定教研室刘圣金副教授鉴定为正品。苯甲酰乌头原碱(批号JBZ-1741)、苯甲酰次乌头原碱(批号JBZ-0039)、苯甲酰新乌头原碱(批号JBZ-0032)、次乌头碱 (批号JBZ-0161)、新乌头碱(批号JBZ-1314)、乌头碱(批号JBZ-1270) 对照品均购自南京金益柏生物科技有限公司,纯度≥98%。95%乙醇及D101、AB-8 大孔树脂均购自南京晚晴化玻仪器有限公司;HPD-826、XDA-5、LS-303 大孔树脂均购自郑州和成新材料科技有限公司。乙腈、甲醇为色谱纯,均购自美国Tedia 公司;其余试剂均为分析纯;水为超纯水。
2 方法与结果
2.1 样品溶液制备
2.1.1 上样液 取黑顺片中粉(过280 μm 筛) 约200 g,加4 倍量85%乙醇浸泡24 h,滤过,滤液备用,药渣用4倍量85%乙醇作为溶剂,加热回流提取4 次,每次2 h,药渣用少量溶剂洗涤,合并浸渍液、提取液、洗涤液,回收乙醇浓缩至无醇味,加蒸馏水制成质量浓度为1 g/mL 的母液,上柱时根据试验设计加水稀释得上样液。
2.1.2 洗脱液 取经预处理的D101 型大孔树脂约10 g,分别装入同样规格的玻璃层析柱中,取0.16 g/mL 上样液50 mL,上柱,体积流量1.5 mL/min,5 BV 蒸馏水清洗杂质,体积流量5 mL/min,8 BV 80%乙醇以1.5 mL/min 体积流量洗脱,收集洗脱液,回收乙醇,蒸馏水定容至50 mL,即得。
2.2 总生物碱含量测定
2.2.1 对照品溶液制备 取乌头碱对照品适量,精密称定,加0.01 mol/L 盐酸制成每1 mL 含0.111 2 mg 该成分的溶液,即得。
2.2.2 线性关系考察 根据文献[12] 方法,以对照品质量浓度为横坐标(X),吸光度为纵坐标(A) 进行回归,得方程为A=0.002 8X-0.002 6 (r=0.999 0),在22.24~133.44 μg/mL 范围内线性关系良好。
2.2.3 测定方法 精密吸取“2.1” 项下上样液、洗脱液各10 mL,置于分液漏斗中,浓氨水调节pH 至10~11,二氯甲烷提取3 次,每次10 mL,合并二氯甲烷层,残渣加0.01 mol/L 盐酸至10 mL,精密吸取3 mL至分液漏斗中,按“2.2.2” 项下方法测定吸光度,计算总生物碱质量浓度。
2.2.4 方法学考察
2.2.4.1 精密度试验 精密称取黑顺片中粉20 g,按“2.1” 项下方法制备样品溶液,按“2.2.3” 项下方法测定6 次,测得吸光度RSD 为0.06%,表明仪器精密度良好。
2.2.4.2 重复性试验 精密称取黑顺片中粉20 g,按“2.1” 项下方法平行制备洗脱液6 份,按“2.2.3” 项下方法测定,测得生物碱含量RSD 为0.46%,表明该方法重复性良好。
2.2.4.3 稳定性试验 精密称取黑顺片中粉20 g,按“2.1” 项下方法制备样品溶液,于0、0.5、1、2、4、8、12、24 h 按“2.2.3” 项下方法测定,测得吸光度RSD 为0.12%,表明溶液在24 h 内稳定性良好。
2.2.4.4 加样回收率试验 取总生物碱含量已知的同一份上样液25 mL,共6 份,精密加入100% 水平乌头碱对照品,按“2.1” 项下方法制备样品溶液,按“2.2.3” 项下方法测定,计算回收率。结果,平均加样回收率为99.42%,RSD 为0.94%。
2.3 单、双酯型生物碱含量测定
2.3.1 色谱条件 Waters XBridge-C18色谱柱(4.6 mm×250 mm,5 μm);流动相乙腈 (A)-5 mmol/L 醋酸铵(B),梯度洗脱(0~12 min,2%A;12~40 min,2%~28%A;40~60 min,28%~45% A;60~62 min,45%~2% A);体积流量1.0 mL/min;柱温30 ℃;检测波长235 nm;进样量10 μL,色谱图见图1。各生物碱与其相邻色谱峰的分离度均大于1.5,理论塔板数按苯甲酰新乌头原碱峰计,应不低于3 000[1]。

图1 各生物碱HPLC 色谱图
2.3.2 对照品溶液制备 取各对照品适量,精密称定,加乙腈制成每1 mL 分别含苯甲酰乌头原碱399.623 μg、苯甲酰次乌头原碱39.960 μg、苯甲酰新乌头原碱12.052 μg、新乌头碱7.980 μg、次乌头碱32.034 μg 的溶液,即得。
2.3.3 供试品溶液制备 按“2.1” 项下方法制备样品溶液,14 000 r/min 离心10 min,取上清液,即得。
2.3.4 方法学考察
2.3.4.1 线性关系考察 取“2.3.2” 项下对照品溶液适量,乙腈依次稀释2 倍,在“2.3.1” 色谱条件下进样测定。以对照品质量浓度为横坐标(X),峰面积为纵坐标(Y) 进行回归,结果见表1,可知各生物碱在各自范围内线性关系良好。
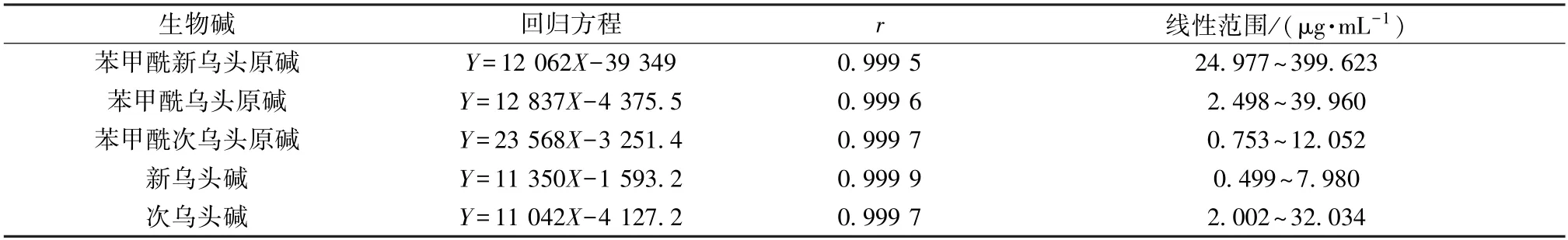
表1 各生物碱线性关系
2.3.4.2 精密度试验 精密吸取“2.3.2” 项下对照品溶液适量,在“2.3.1” 项色谱条件下进样测定6 次,测得苯甲酰乌头原碱、苯甲酰次乌头原碱、苯甲酰新乌头原碱、新乌头碱、次乌头碱峰面积RSD 分别为2.16%、2.69%、4.48%、3.50%、3.79%,表明仪器精密度良好。
2.3.4.3 重复性试验 按“2.3.3” 项下方法制备供试品溶液6 份,在“2.3.1” 项色谱条件下进样测定,测得苯甲酰乌头原碱、苯甲酰次乌头原碱、苯甲酰新乌头原碱、新乌头碱、次乌头碱含量RSD 分别为1.74%、3.11%、4.16%、3.41%、2.25%,表明该方法重复性良好。
2.3.4.4 稳定性试验 精密吸取同一份供试品溶液,于0、0.5、1、2、4、8、12、24 h 在“2.3.1” 项色谱条件下进样测定,测得苯甲酰乌头原碱、苯甲酰次乌头原碱、苯甲酰新乌头原碱、新乌头碱、次乌头碱峰面积RSD 分别为1.37%、50 mL。2.09%、4.83%、2.45%、2.85%,表明溶液在24 h 内稳定性良好。
2.3.4.5 加样回收率试验 取各生物碱含量已知的供试品溶液6 份,加入100%对照品溶液适量,在“2.3.1” 项色谱条件下进样测定,计算回收率。结果,苯甲酰乌头原碱、苯甲酰次乌头原碱、苯甲酰新乌头原碱、新乌头碱、次乌头碱平均加样回收率分别为92.21%、99.75%、100.25%、102.34%、98.23%,RSD 分别为2.14%、3.30%、3.98%、4.96%、3.40%。
2.3.5 样品含量测定 取黑顺片中粉适量,按“2.3.3”项下方法制备供试品溶液,在“2.3.1” 项色谱条件下进样测定,平行2 次,计算含量。
2.4 树脂筛选
2.4.1 预处理 用95%乙醇溶液浸泡树脂24 h 以保证其充分溶胀,湿法装柱,95%乙醇淋洗至洗脱液与水混合(比例为1 ∶2) 不呈白色浑浊为止,再用去离子水洗至无醇味,湿态保存备用。
2.4.2 静态吸附试验 称取预处理后表面干燥的树脂约1 g,置于150 mL 锥形瓶中,加入10 mg/mL 总碱溶液10 mL,室温下120 r/min 振荡吸附24 h,测定吸附后溶液中总生物碱含量,计算吸附率,公式为吸附率=[(吸附前总生物碱含量-吸附后总生物碱含量)/吸附前总生物碱含量] ×100%,结果见表2。

表2 树脂静态吸附-解吸附性能测定结果
2.4.3 静态解吸附试验 将上述吸附生物碱的树脂分离,少量水洗涤后除去水分,加入10 mL 70%乙醇,室温下120 r/min振荡脱附24 h,测定洗脱液中总生物碱含量,计算解吸率,公式为解吸率=[洗脱液中总生物碱含量/(吸附前总生物碱含量-吸附后总生物碱含量)] ×100%,结果见表2。
由此可知,D101、LS-303 树脂吸附率均达到80%以上,并且前者吸附率、解吸率更大。因此,选用D101 树脂进行下一步分离纯化。
2.5 单因素试验
2.5.1 上样量(动态吸附曲线) 按“2.4.1” 项下方法湿法装柱后,取0.1 g/mL 样品溶液,每10 mL 收集1 个流分,按“2.2.3” 项下方法测定总生物碱含量,以流出液体积为横坐标(X),流出液中总生物碱浓度为纵坐标(Y),绘制动态吸附的泄露曲线[7-8]。由图2A 可知,当上样液体积为10 mL 时,附子生物碱开始少量泄露,随着上样液体积的增大,流出液中总生物碱浓度呈上升趋势;达到50 mL时,随着上样体积增加,流出液总生物碱质量浓度趋于平缓,可能是由于大孔树脂吸附达到饱和,故确定上样量为

图2 单因素试验结果
2.5.2 上样液质量浓度 按“2.4.1” 项下方法湿法装柱后,分别取0.05、0.1、0.25、0.5 g/mL 样品溶液各100、50、20、10 mL,调节pH 值至5.0,上柱,体积流量1.5 mL/min,蒸馏水5 BV 清洗杂质,体积流量5 mL/min,收集流出液,按“2.2.3” 项下方法测定总生物碱含量,计算吸附率。由图2B 可知,当上样液质量浓度为0.1 g/mL 时,吸附量达到最高,吸附率最大,为98.8%;随着其进一步升高,吸附量下降,可能是发生了絮凝和沉淀现象,故选择上样液质量浓度为0.1 g/mL。
2.5.3 上样液pH 值 按“2.4.1” 项下方法湿法装柱后,取0.1 g/mL 样品溶液50 mL,调节pH 值至3.0、5.0、7.0、9.0,上柱,体积流量1.5 mL/min,收集流出液,按“2.2.3” 项下方法测定总生物碱含量,计算吸附率。由图2C 可知,pH 对于吸附效果影响不大,原液pH 为3.49,可直接上样,无需调pH 值。
2.5.4 洗脱液体积分数 按“2.4.1” 项下方法湿法装柱后,取0.1 g/mL 样品溶液50 mL,上柱,体积流量1.5 mL/min,蒸馏水5 BV 清洗杂质,体积流量5 mL/min,分别用10%、30%、60%、80%乙醇各10 BV 洗脱,收集洗脱液,回收乙醇,蒸馏水定容,按“2.2.3” 项下方法测定总生物碱含量,计算解吸率。由图2D 可知,随着洗脱液体积分数增加解吸率不断升高,为80%时达到最大值,故选择其作为最优洗脱液体积分数。
2.5.5 洗脱液用量(动态洗脱曲线) 按“2.4.1” 项下方法湿法装柱后,取0.1 g/mL 样品溶液50 mL,上柱,体积流量1.5 mL/min,蒸馏水5 BV 清洗杂质,体积流量5 mL/min,80%乙醇10 BV 洗脱,每1 BV 收集1 个流分,按“2.2.3” 项下方法测定各流分中总生物碱含量,绘制动态解吸附曲线。由图2E 可知,洗脱液中总生物碱含量随乙醇洗脱液体积增加呈先增加后减少的趋势,为3 BV 时达到最大值;随着其继续增大,生物碱含量开始下降,6 BV 后已经很少,表明总生物碱接近洗脱完全。因此,最佳洗脱体积为6 BV,但因兼顾生物碱得率,还需进一步考察洗脱液用量。
2.6 星点设计-效应面法
2.6.1 设计方案 根据单因素试验结果,选择上样液质量浓度(A)、乙醇体积分数(B)、乙醇洗脱量(C) 作为影响因素,根据星点设计原理,采用三因素五水平试验,水平用代码值-α、-1、0、1、α 表示,因素水平见表3。取各质量浓度样品溶液50 mL,以1.5 mL/min 体积流量上样,按表3 进行洗脱,回收乙醇,蒸馏水定容,采用酸性染料比色法测定洗脱液中总生物碱含量,HPLC 法测定双、单酯型生物碱总量,并计算解吸率、纯度,公式为纯度=[样品液中总生物碱的含量/样品液固含量] ×100%,其中样品液固含量参照2020 年版《中国药典》 通则项下3101 固体总量测定法[13]进行测定。再将表3 中的总生物碱(以乌头碱计)、双酯型生物碱和单酯型生物碱总量的数据进行“归一化”,运用Hassan 法分别对各指标进行数学转换求“归一值” (d),公式为d=(Yi-Ymin)/(Ymax-Ymin),计算总评归一值(OD),公式为OD=(d1×d2×d3)1/3,结果见表3。

表3 试验设计与结果
2.6.2 数据处理 采用Design-Expert 8.0 软件对表3 中数据进行多元线性回归、二项式拟合,得多元线性回归方程为Y=-1.676-5.566×10-3A+0.026 3B+0.058 3C(r=0.732 1,P<0.01),虽然P值通过检验,但相关系数过小,模型拟合度不高,预测性较差,而二项式拟合方程为Y=-7.533+ 10.00A+ 0.178 8B-3.866 × 10-3C-0.035AB-0.022 9AC+6.717×10-4BC-17.02A2-1.079×10-3B2+3.944×10-3C2(r=0.944 6,P<0.01),方差分析见表4。

表4 方差分析
由此可知,模型P<0.01,表明该拟合模型具有显著性,拟合度R2=0.944 6,失拟项P>0.05,说明拟合度很高,误差小,模型可靠;校正决定系数=0.894 7,说明模型可信度较高,能解释89.47%响应值的变化,能较好地拟合真实效应面;一次项B对OD 值有极显著影响(P<0.01),C对OD 值有显著影响(P<0.05),二次项A2、B2对OD 值有极显著影响(P<0.01),交互项AB、AC、BC均不显著(P>0.05)。
2.6.3 工艺预测 根据上述二次多项式拟合模型,分别固定一个自变量为中间值,绘制因变量解吸率随自变量变化的三维效应面图、二维等高线图,见图3。由此可知,乙醇体积分数、乙醇洗脱量的曲线均较陡峭,说明乙醇体积分数对OD 值的影响最显著。用Point Prediction 进行预测分析,可知最优工艺为上样液质量浓度0.16 g/mL,乙醇体积分数80%,乙醇洗脱量8 BV,OD 值为0.941 3。

图3 各因素响应面图
2.6.4 验证试验 取0.16 g/mL 样品溶液50 mL,上柱,体积流量1.5 mL/min,蒸馏水5 BV 清洗杂质,体积流量5 mL/min,8 BV 80%乙醇以1 mL/min 体积流量洗脱,采用酸性染料比色法测定洗脱液中总生物碱含量,HPLC 法测定双、单酯型生物碱总量,并计算解吸率、纯度,平行3 次,取平均值,并计算OD 值。结果,与预测值0.941 3 偏差-7.30%,表明所得拟合方程可较好地描述指标与因素之间的关系,模型合理可靠,预测性好,可进行提取纯化,见表5。

表5 验证试验结果(n=3)
3 讨论与结论
在确定评价指标时,在上柱因素考察过程中,考虑到损失率,在饱和时即停止上样,上样流出液中生物碱类成分量较低,无法测定单一生物碱的量,因此以总生物碱的吸附率为指标优选上柱参数。在洗脱因素考察过程中,本实验主要是研究总生物碱的富集纯化工艺,因此总生物碱在洗脱工艺中占较大的比重,而单一成分在洗脱工艺研究中也占有一定的权重,双酯型生物碱、单酯型生物碱为附子的主要有效成分,因此选用总生物碱、双酯型与单酯型生物碱总量的解吸率和纯度为指标总评归一值进行洗脱参数优选[14-17]。
本实验对色谱条件进行了优化,考察了Waters XBridge-C18色谱柱(4.6 mm×250 mm,5 μm)、Angilent ZORBAXSB C18色谱柱(4.6 mm×250 mm,5 μm)、Thermo Acclaim TM 120 C18色谱柱(4.6 mm×250 mm,5 μm),比较了流动相乙腈-四氢呋喃 (25 ∶15)-0.1 mol/L 醋酸铵、乙腈-40 mmol/L醋酸铵、乙腈-5 mmol/L 醋酸铵对色谱峰分离度的影响,并比较了不同检测波长下色谱峰数量及峰高,最终确定为Waters XBridge-C18色谱柱 (4.6 mm×250 mm,5 μm),乙腈-5 mmol/L 醋酸铵梯度洗脱,此时各成分分离度、基线平稳、峰形均良好。
本研究通过单因素考察并结合星点设计-效应面法筛选附子生物碱最佳纯化工艺,采用紫外吸收光度法及HPLC对生物碱类成分进行含量测定,结合纯度进行综合评价,经验证本纯化工艺稳定可行,可为附子的综合利用提供依据。
猜你喜欢
中国饲料(2021年17期)2021-11-02
中成药(2018年11期)2018-11-24
中成药(2018年2期)2018-05-09
中成药(2017年12期)2018-01-19
中成药(2017年7期)2017-11-22
中成药(2017年8期)2017-11-22
广东饲料(2016年5期)2016-12-01
中成药(2016年4期)2016-05-17
合成化学(2015年10期)2016-01-17
中国当代医药(2015年24期)2015-03-01
