
固相萃取-二维液相色谱法同时测定婴儿配方乳粉中VA、VD和VE
2023-09-09 02:04:34宁霄金绍明刘彤彤赵梅曹进
化学分析计量 2023年8期
宁霄,金绍明,刘彤彤,赵梅,曹进
(中国食品药品检定研究院,北京 100050)
二维液相色谱通过两个具有不同分离机制的色谱柱串联组合,将目标物从一维柱转移到二维柱进一步分析。由于二维液相色谱可以去除复杂样品基质中的干扰物质,实现多种组分同步检测,提高检测灵敏度,近年来已成为复杂样品制备和检测的主要方法[7]。现有二维分离系统检测乳粉中脂溶性维生素的报道中,样品预处理多参照国家标准采用皂化、有机溶剂萃取等,方法精密度和回收率有待提高[8]。也有利用在线净化与二维液相色谱联用对相关组分进行定量分析[9‒11],但仪器成本高,不利于推广,且分析准确度有待提高。在利用固相萃取柱对乳粉皂化液进行处理的报道中[12‒13],VD的检测需要经正相硅胶柱净化,方法操作复杂,且未实现3种维生素同步测定。
笔者采用正交试验对皂化条件进行优化,最大程度释放目标组分。采用固相萃取法取代GB 5009.82—2016[14]中的有机溶剂萃取法,并使用二维液相色谱法对样品溶液进行分析,省去正相制备步骤,简化样品预处理流程,实现一次进样同时完成VA、VD和VE含量的测定,可以满足实验室大批量样品快速分析要求,也可用于乳粉中VA、VD和VE基质标准物质定值及产品质量控制。
1 实验部分
1.1 主要仪器与试剂
液相色谱仪:U3000 型双三元液相色谱(2DLC)系统,配有DAD检测器,美国赛默飞世尔公司。
电子天平:XPE205 型,感量为0.01 mg,瑞士梅特勒-托利多公司。
维生素专用固相萃取柱:TUP-E76-2060 型,规格为20 g/(60 mL),天津诚轴生物科技有限公司。
VA(视黄醇)对照品:质量分数不小于98.7%,相对扩展不确定度为1.77%,k=2,美国西格玛公司。
VD3对照品:质量分数不小于99.8%,相对扩展不确定度为0.3%,k=2,德国DR.E公司。
(二)强化信息收集 掌握市场信息是产业发展的基础,强化生猪生产信息收集是把握市场、科学决策的保障。要迅速启动区畜牧局、基层站、业主微机网络联网管理,广泛收集国内外、重庆市内外及周边县市生猪市场行情,及时发布和交流市场信息,实现网上资源共享,做到及时得到市场信息,如市场价格、供求关系、疫情动态等。适时调整养殖数量、品种,把握市场主动权,提高业主抗市场风险能力,防止盲目发展导致价格下降造成损失,挫伤业主养殖积极性。
VE(D-α-生育酚)对照品:质量分数不小于98.2%,相对扩展不确定度为2.0%,k=2,德国DR.E公司。
抗坏血酸、2,6-二叔丁基对甲酚(BHT):分析纯,国药集团化学试剂有限公司。
无水乙醇、乙腈、甲醇、正己烷:色谱纯,德国默克公司。
1.2 仪器工作条件
一维分析泵:2D-LC系统的右泵;一维分析柱:FOODKIT1 ADE-C18色 谱 柱(150 mm×3.0 mm,3 μm,美国赛默飞世尔公司);一维分析流动相:乙腈-甲醇-水体系,流量为0.5 mL/min,梯度洗脱,洗脱程序见表1;二维分析泵:2D-LC 系统的左泵;二维分析柱:FOCDKIT1 & 2ADE-C18色谱柱(100 mm×4.6 mm,2.6 μm,美国赛默飞世尔公司);二维分析流动相:乙腈-甲醇-水体系,流量为0.8 mL/min,梯度洗脱,洗脱程序见表1;柱温:30 ℃;检测波长:VA325 nm、VD264 nm、VE296 nm;进样体积:20 μL。阀门切换时间见表2。

表1 梯度洗脱程序

表2 阀门切换时间
1.3 实验步骤
1.3.1 样品处理
精密称取婴儿配方乳粉5 g 于250 mL 锥形瓶中,加入20 mL 温水(50 ℃)溶解后,加入抗坏血酸1.0 g、BHT 0.1 g、无水乙醇50 mL,质量分数为50%的KOH 溶液15 mL,于65 ℃水浴震荡皂化50 min,将皂化液冷却后用水-乙醇(体积比1∶1)混合液转移定容至100 mL,摇匀,在8 000 r/min 下离心5 min,取20 mL 上清液转移至维生素专用固相萃取柱,用100 mL 正己烷(含BHT 质量浓度为50 mg/L)以约5 mL/min 的流量洗脱,洗脱液收集至旋蒸瓶中,于40 ℃旋蒸仪旋蒸至约2 mL 时转移至离心管中,用氮气吹干,准确加入2 mL 甲醇复溶,经0.22 μm 微孔滤膜过滤,上机分析。
1.3.2 样品测定
试样液经二维液相色谱仪分析,测得峰面积,采用外标法通过上述标准曲线计算其浓度。
2 结果与讨论
2.1 样品净化方法优化
为了减少这三种类脂溶性维生素在运输和储存过程中的流失,婴儿配方乳粉制备工艺中需要通过乙酸酯化并微胶囊化处理,样品需经过皂化处理,才能将样品中的VA、VD、VE溶解出来。GB 5009.82—2016采用有机溶剂萃取法净化皂化液,该方法易出现两相界面乳化现象,导致目标组分从皂化液向萃取液转移过程中损失,且光敏性强的目标组分在萃取液浓缩过程中极易见光分解,从而导致回收率较低。采用固相萃取柱净化皂化液,可以减少复杂操作引入的待测组分损失。考虑到乳粉中VD含量低,为实现与VA、VE的同步检测,需利用大容量的固相萃取柱富集。经过对比试验,最终选择规格为20 g/(60 mL)的TUP-E76-2060维生素专用固相萃取柱。
2.2 皂化时间和温度的选择
为全面考察2个影响因素,以皂化时间(A)和皂化温度(B)2 个因素,每个因素3 水平,设计正交试验,考察最佳提取条件,试验水平见表3。分别选择皂化时间为40、50、60 min,皂化温度为50、65、80 ℃,考察3 种维生素的平均回收率(平行测定3次,求平均值)。正交试验方案及试验对应维生素的回收率见表4,统计结果见表5。

表3 正交因素-水平表
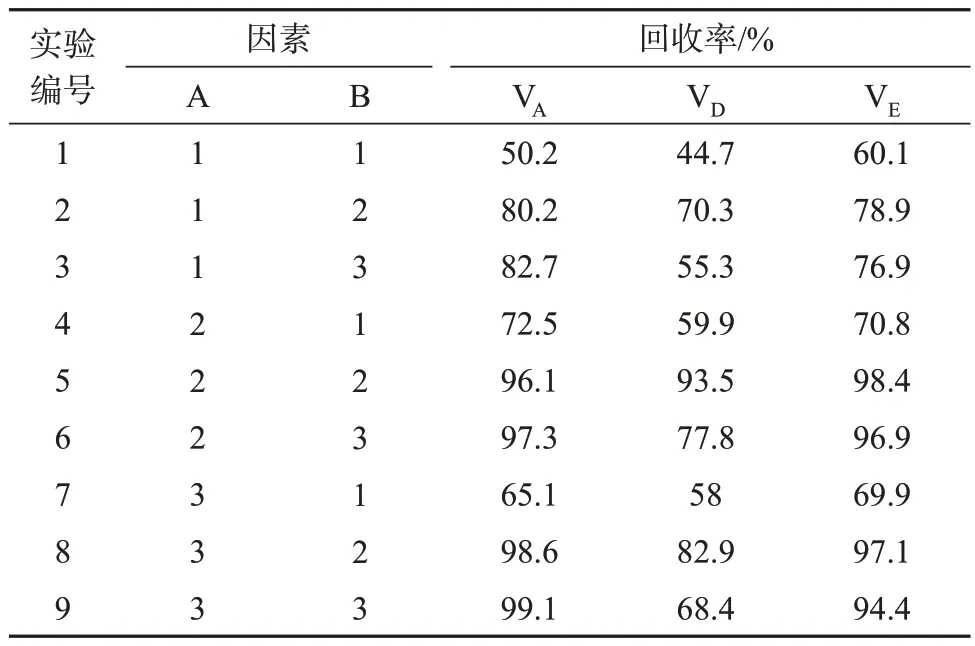
表4 正交试验方案及试验对应维生素的回收率
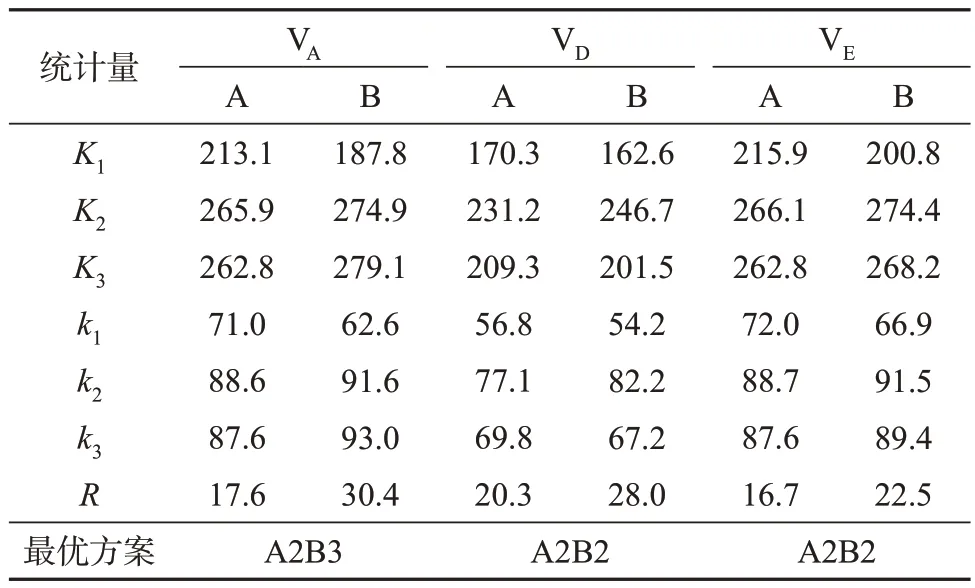
表5 正交试验回收率统计结果
由表4 可知,当皂化温度为50 ℃时,样品皂化不完全导致各种维生素回收率相对较低;当皂化温度升高至65 ℃时,维生素回收率明显上升;继续提高皂化温度至80 ℃,VD回收率明显下降。原因可能是VA和VE均耐高温,表面包被被破坏,其构型从酯型更加充分地转化为游离醇型,致使萃取效率更高。而VD随温度升高发生少量氧化损失,导致回收率下降。综合以上情况,确定皂化温度为65 ℃。固定皂化温度为65 ℃,当皂化时间从40 min 延长至50 min时,3种维生素回收率呈现上升趋势;继续延长皂化时间VA和VE回收率变化不明显,提取率前者略有提高,后者略有下降,VD情况相反,在80 ℃下皂化时回收率明显降低。
由表5中3种维生素回收率的R值可以看出,皂化温度的影响大于皂化时间。经过运算可得出每个因素各水平之和K1、K2、K3,综合各因素K值和直观分析,在取样量5 g时,A2B2为VD和VE最佳提取条件,虽然理论上VA提取效果最佳的皂化条件为A2B3,但改善效果不明显,反而大幅影响了VD的回收率,因此最佳提取条件确定为A2B2。
表6 为正交试验方差分析结果,进一步反映了皂化时间(A)、皂化温度(B)两个因素对目标物回收率影响的程度(P<0.05)。由表6可知,皂化时间和皂化温度是该实验的显著性因素,故确定其提取回收率最大的皂化条件:温度65 ℃,时间50 min。

表6 正交试验方差分析结果
2.3 乙醇溶液添加量的选择
由于三种维生素脂溶性较强,因此加入乙醇以增加溶解度,当乙醇含量较低时,目标物溶解度较小,极易形成乳浊液,而乙醇含量过高则会导致目标组分在固相萃取小柱中不能完全保留,乙醇使用量对目标物回收率的影响如图1所示。

图1 乙醇不同使用体积对应的目标物回收率
2.4 洗脱液使用量的选择
实验使用正己烷作为洗脱液。当皂化液上样量为20 mL 时,分别选择使用正己烷50、80、90、100 mL,考察正己烷的使用量对待测组分的洗脱效果,结果如图2 所示。由图2 可知,使用100 mL 正己烷可将3种维生素完全洗脱下来。
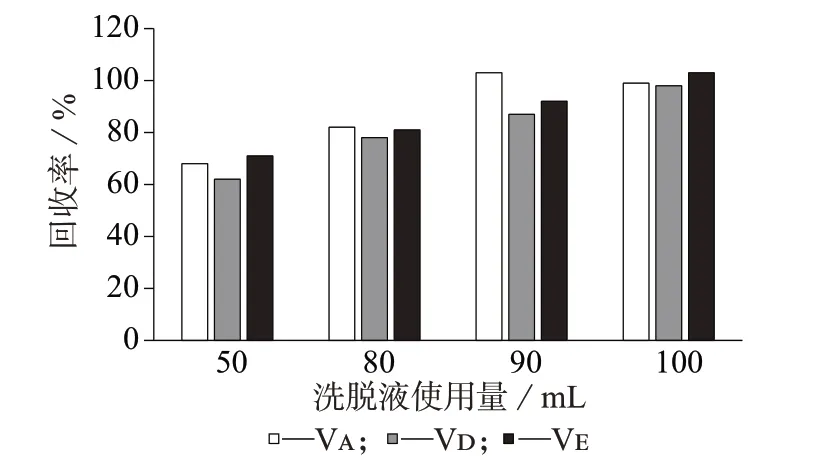
图2 洗脱液不同用量对应的目标物回收率
2.5 线性范围、检出限和定量限
配制三种维生素目标物的系列标准工作溶液,按1.2仪器工作条件测定,以目标物质量浓度为横坐标x轴、色谱峰面积为纵坐标y轴,绘制标准工作曲线,计算线性方程和相关系数。
以加标样品色谱峰响应值为3倍信噪比的添加浓度为方法检出限,以10倍信噪比的添加浓度为方法定量限。
三种维生素在婴儿配方乳粉中的线性范围、线性方程、相关系数、检出限和定量限见表7。

表7 线性范围、线性方程、相关系数、检出限和定量限
由表7 可见,VA质量浓度在0.005~5 μg/mL 范围内相关系数为0.999 6,VD质量浓度在0.01~10 μg/mL 范围内相关系数为0.999 8,VE质量浓度在0.5~500 μg/mL 范围内相关系数为0.999 9,表明各目标物色谱峰面积与对应标准溶液的质量浓度线性关系良好。
2.6 加标回收试验
采用已知目标组分含量的实际样品进行加标回收试验。在样品中添加适量待测组分标准溶液,使添加水平分别为定量限的1、5、10 倍。每个添加水平进行6次平行试验,试验结果见表8,空白样品、混合标准溶液及加标样品溶液色谱图见图3。由表8可知,3 种维生素的平均加标回收率为75.7%~104.5%,相对标准偏差均小于5.7%,证明此样品预处理方法及二维液相色谱方法具有较好的回收率和重现性,满足GB/T 27417—2017附录A检测方法确认的技术要求[15],适用于婴儿配方乳粉中3 种脂溶性维生素的同时测定。
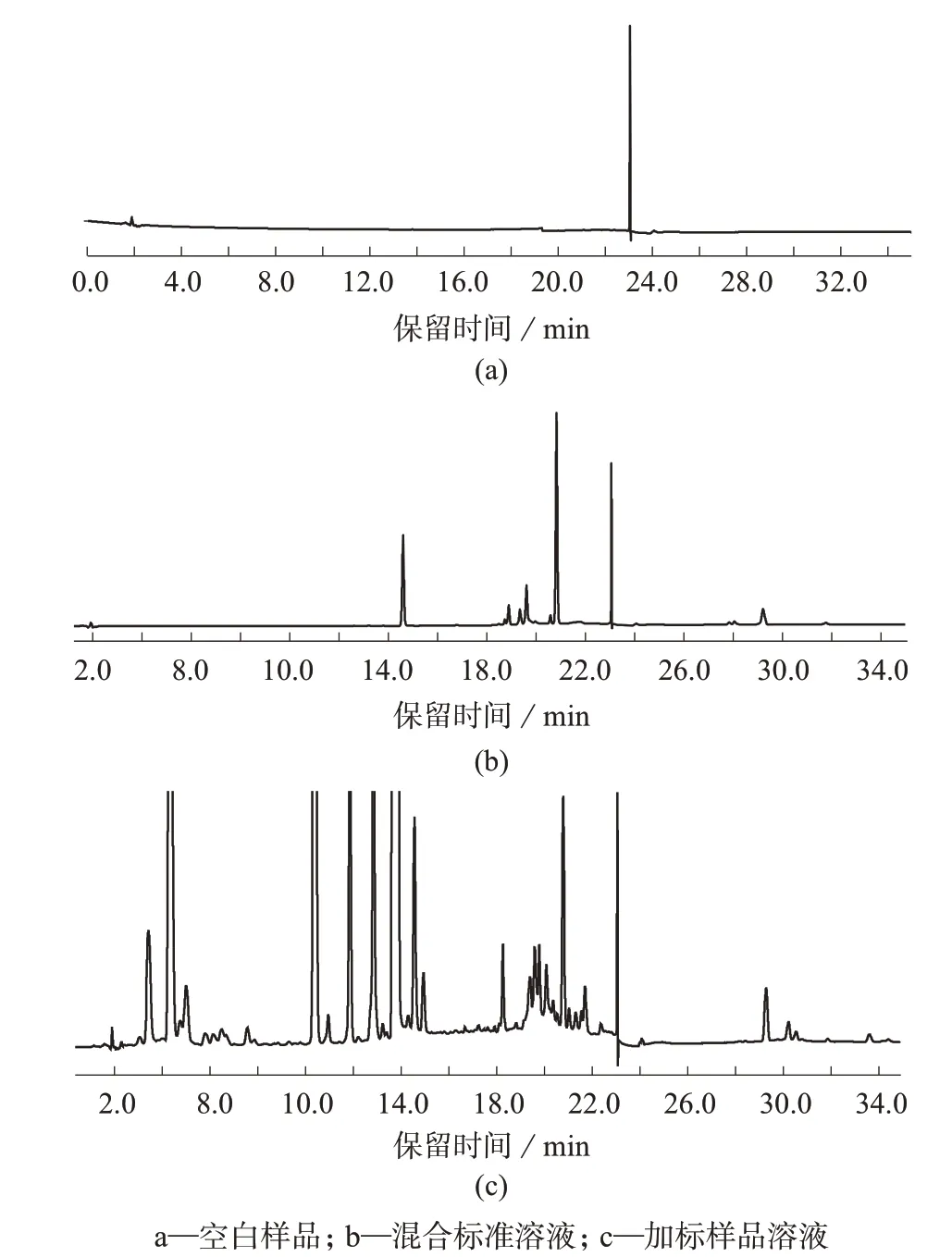
图3 空白样品、混合标准溶液及加标样品溶液色谱图
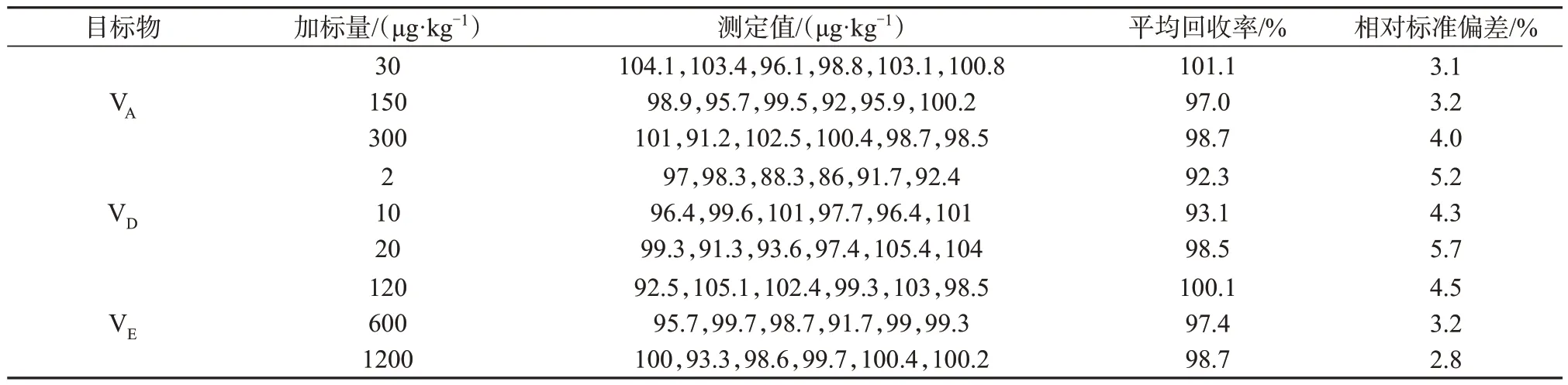
表8 婴儿配方乳粉中3种维生素的加标回收试验结果
2.7 方法比对试验
采用已知浓度的乳粉中VA、VD和VE基质标准物质作为样品,加入与其浓度相当的标准溶液,分别用国家标准方法和该方法进行测定,两种方法检出限、定量限、回收率和测定值的相对标准偏差见表9。结果表明,与现行国标方法比较,该方法的准确度及精密度均有改善,分析灵敏度均有不同程度的提升,其中VA的定量限提高了30倍,VD的回收率提升了1.6倍。
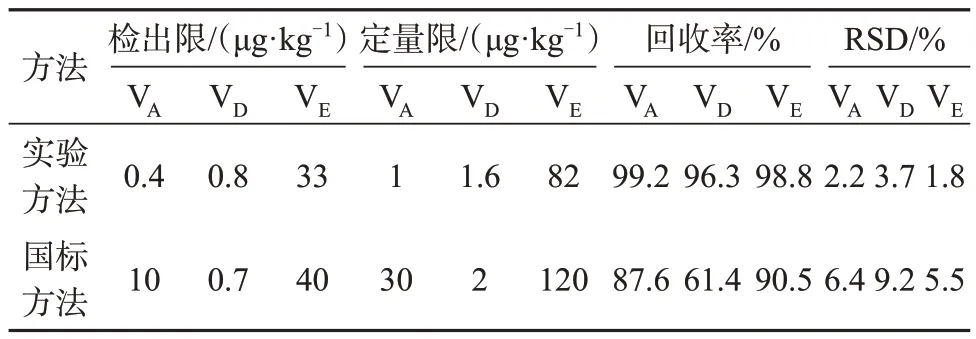
表9 比对试验结果
3 结语
建立了婴儿配方乳粉中VA、VD和VE含量同步分析的二维液相色谱方法,该方法利用固相萃取方式,省去了有机溶剂液液萃取的繁杂步骤,实现了三种脂溶性维生素的同步检测。该方法操作简便,准确稳健,灵敏度高,可满足实验室大量、快速分析的需求。
猜你喜欢
食品科学(2023年4期)2023-03-06 12:49:32
煤气与热力(2021年12期)2022-01-19 05:19:30
食品安全导刊(2021年21期)2021-08-30 08:21:58
成都大学学报(自然科学版)(2021年1期)2021-05-22 01:31:24
中国乳业(2020年12期)2020-04-12 01:12:46
中成药(2018年8期)2018-08-29 01:28:26
中成药(2018年2期)2018-05-09 07:20:09
锻压装备与制造技术(2018年1期)2018-03-28 03:49:36
食品安全导刊(2014年8期)2014-10-21 14:57:42
中国酿造(2014年9期)2014-03-11 20:21:13
