
液相色谱质谱法测定鸡肉中尼卡巴嗪残留标志物残留量的不确定度评定
2023-09-08 10:44张丽芳康俊杰张建雄郑百芹
中国动物传染病学报 2023年3期
张丽芳,康俊杰,张建雄,郑百芹,刘 洋
(1.唐山市食品药品综合检验检测中心,唐山 063000;2.河北省农产品质量安全检测技术创新中心,唐山 063000;3.唐山市功能性农产品产业技术研究院,唐山 063000;4.农业农村部农产品质量监督检验测试中心(唐山),唐山 063000))
尼卡巴嗪(nicarbazin)又名球虫净,是一种广谱、高效和性能稳定的抗球虫药,可有效预防和治疗鸡等禽类的球虫病。它预防效果好,耐药性极慢,安全性高,有促生长作用,被广泛应用于鸡的养殖环节[1-2]。尼卡巴嗪由4,4-二硝基均二苯脲(4,4'-Dinitrocarbanilide,DNC)与2-羟基-4,6-二甲基嘧啶(2-hydroxy-4,6-dimethylpyrimidine,HDP)两种化合物组成,两者均由家禽消化道吸收,并广泛分布于组织及体液中。DNC通过粪便排出,代谢缓慢,HDP以尿液排出,代谢消除迅速[3]。国内外均以DNC作为尼卡巴嗪在鸡组织中的残留标志物[4]。GB31650-2019[5]规定尼卡巴嗪残留标志物在鸡的所有组织中最高残留限量为200 μg/kg。鸡组织中残留的DNC经烹饪加热后可诱导产生对硝基苯胺[6],此化合物具有高毒性。尼卡巴嗪在禽类组织中不同程度的残留会对人体健康产生潜在危害[7-8]。
测量不确定度定义为表征合理地赋予被测量之值分散性的非负参数。它是定量阐明检测结果品质优良的重要指标[9-11],同时可分析和评定影响测量结果准确性的各个分量,充分掌握关键因素,提高结果的准确性和可靠性。不确定度作为合格判定的重要参考依据,将最大限度地减少最大残留限量附近残留水平合规决定中可能存在的争议[12-13]。CNASGL006:2019《化学分析中不确定度的评估指南》[14]改进了当测量结果接近于零时的不确定度评定,完善了带有测定不确定的结果的符合性判定指南,使实验室对提供测量不确定度的要求和方法日趋明确。目前兽药残留量的测量不确定评定方法仍不成熟[15-19],存在一定的局限:如方法回收率未作为评定的考虑因素,忽略试剂的膨胀,量器在计算不确定度过程中选择矩形分布、三角分布或正态分布计算方式不明确等。本文以GB29690-2013[20]《食品安全国家标准 动物性食品中尼卡巴嗪残留标志物残留量的测定 液相色谱-串联质谱法》为检测方法,采用液相色谱质谱法对鸡肉中尼卡巴嗪残留标志物残留量进行测定,依据JJF1135 -2005[21]《化学分析测量不确定度评定》、JJF1059.1-2012[22]《测量不确定度评定与表示》和CNAS- GL006-2019,综合考虑各个因素,进行测量不确定的评定,建立鸡肉中尼卡巴嗪残留标志物残留量不确定度评定的数学模型,分析影响测量结果准确性的各关键因素,加强质量控制,为科学评价药物残留量结果提供依据。
1 材料与方法
1.1 材料与仪器 标准物质4,4'-二硝基均二苯脲(含量100 μg/mL,不确定度5%)和4,4'-二硝基均二苯脲-D8(含量100 μg/mL,不确定度5%),均购自天津阿尔塔有限公司;甲醇(色谱纯)、乙腈(色谱纯)、正己烷(色谱纯)购自德国Merck;无水硫酸钠(分析纯)购自天津福晨化学试剂;试验用鸡肉样品购自当地超市。液相色谱-串联质谱仪(Waters Xevo TQ)购自美国Waters公司;电子天平(HZ2004A)购自常州市衡正电子仪器有限公司;台式离心机(D37520)购自德国Thermo公司;氮吹仪(TTL-DC)购自北京同泰联科技发展有限公司。
1.2 试液配制 75%甲醇水:量取甲醇75 mL,用水溶解稀释至100 mL;75%甲醇水饱和的正己烷:取75%甲醇水100 mL,加正己烷100 mL,摇匀静置分层,取上层液;4,4'-二硝基均二苯脲贮备液:精密吸取4,4'-二硝基均二苯脲标准物质1.0 mL置于10 mL容量瓶中,用甲醇定容制备成10 μg/mL的储备液;1.2.4 4,4'-二硝基均二苯脲中间液:精密吸取4,4'-二硝基均二苯脲储备液0.1 mL分别置于1 mL容量瓶中,用75%甲醇水定容制备成1 μg/mL的中间液;4,4'-二硝基均二苯脲-D8内标工作液:精密吸取4,4'-二硝基均二苯脲-D8标准物质0.1 mL置于100 mL容量瓶中,用75%甲醇水定容制备成100 ng/mL的内标工作液;标准工作曲线的制备:吸取4,4'-二硝基均二苯脲中间液液0.02 mL 置于10 mL容量瓶中;吸取4,4'-二硝基均二苯脲储备液0.01 mL、0.02 mL、0.05 mL、0.2 mL、0.5 mL置于10 mL容量瓶中,准确移取4,4'-二硝基均二苯脲-D8工作液1.0 mL置各容量瓶中,用75%甲醇水定容,制备成2、10、20、50、200、500 ng/mL的标准曲线,4,4'-二硝基均二苯脲-D8内标浓度10 ng/mL。
1.3 试样提取与净化 称取待测鸡肉2.00±0.02 g,于50 mL离心管中,添加100 ng/mL 4,4'-二硝基均二苯脲-D8内标工作液100 μL,加无水硫酸钠2 g,乙腈8 mL,涡旋0.5 min,超声5 min,5000 ×g离心10 min,取上清液,于40℃氮气吹干,加75%甲醇水饱和的正己烷1 mL,涡旋10 s,再加75%甲醇水溶液1.0 mL,充分涡旋混合,于40℃水浴中静置5 min,2000 ×g离心5 min,取下层清液,滤膜过滤,供液相色谱-串联质谱测定。
1.4 仪器条件 色谱柱:Waters ACQUITY UPLC HSS T3(2.1×100 mm,1.7 μm);柱温:40℃;流速:0.3 mL/min;进样量:5.0 μL;流动相:A水,B乙腈;梯度洗脱:0-0.50 min,10%B;0.50-1.20 min,10%B-90%B;1.20-2.10 min,90%B;2.10-2.20 min,90%B-10%B;2.20-3.20,10%B。
电喷雾离子源;负离子扫描;多反应监测;-3500V电喷雾电压;源温:110℃;雾化温度:350℃;雾化气流速:450L/h;锥孔气流速:50L/h;监测离子:DNC 300.9>136.9(定量),300.9>106.9(定性),DNC-D8内标300.9>141.0。
2 结果
2.1 不确定度来源分析 根据试验检测步骤和建立的数学模型,影响LC-MS/MS测定鸡肉中DNC残留量不确定度的因素有以下几个方面:①标准溶液配制引入的不确定度urel(C);②内标加入体积引入的不确定度urel(N);③称样量引入的不确定度urel(M);④样品前处理过程引入的不确定度urel(E);⑤样品的重复测定引入的不确定度urel(X);⑥标准曲线拟合引入的不确定度urel(Y);⑦回收率测定引入的不确定度urel(fR)。
2.2 标准溶液配制引入的不确定度
2.2.1 标准物质引入的不确定度 依据标准物质证书,4,4'-二硝基均二苯脲的扩展不确定为5%(k=2),其相对标准不确定为0.05/2=0.025,4,4'-二硝基均二苯脲-D8内标的扩展不确定度为5%(k=2),其相对标准不确定为0.05/2=0.025,标准物质引入的相对不确定度为urel(C1)
2.2.2 标准储备液配制引入的不确定度 配制标准储备液时,用1000 μL的移液器移取100 μg/mL的标准物质1000 μL于10 mL容量瓶中,用甲醇定容,配制成10 μg/mL的储备液。该过程不确定度的引入涉及到试剂膨胀、移液器和容量瓶的影响。实验室温度通常在20℃±5℃,甲醇的膨胀系数为0.00118℃-1,服从矩形分布,引入的不确定度为0.00118×5/=0. 00341;1000 μL移液器检定证书给出的扩展不确定度为0.3%(k=2),引入的不确定度为0.003/2=0.00150;A级10 mL容量瓶检定给出的不确定度为0.008 mL(k=2),引入的不确定度为0.008/2/10=0.00040。标准储备液配制引入的不确定度urel(C2)==0.00375。
2.2.3 标准工作曲线配制引入的不确定度 由标准曲线配制过程可知,稀释定容过程使用10 mL容量瓶6次,1 mL容量瓶1次,100 μL可调移液器5次,1000 μL可调移液器2次。
2.2.4 容量瓶引入的不确定度 容量瓶引入不确定度的因素:①容量瓶检定的允许扩展不确定度;②实验室温度波动引起的体积变化。75%甲醇水体积膨胀系数为0.000937℃-1,计算如表1。
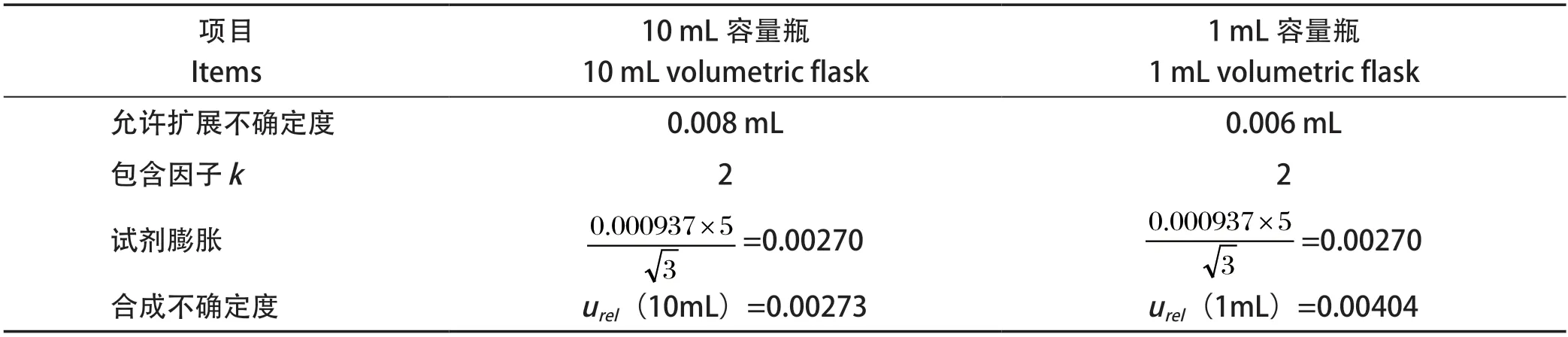
表1 稀释过程中容量瓶引入的不确定度Table 1 Uncertainty of the volumetric flask in the dilution process
2.2.5 可调移液器引入的不确定度 标准工作曲线配制过程中使用100 μL、1000 μL移液器移取不同体积液体,引入不确定度的因素:①移液器本身的允许扩展不确定度;②实验室温度波动引起的体积变化,根据仪器根据实际操作和检定证书,将标准工作曲线稀释过程可调移液器引入的相对不确定度计算如表2。
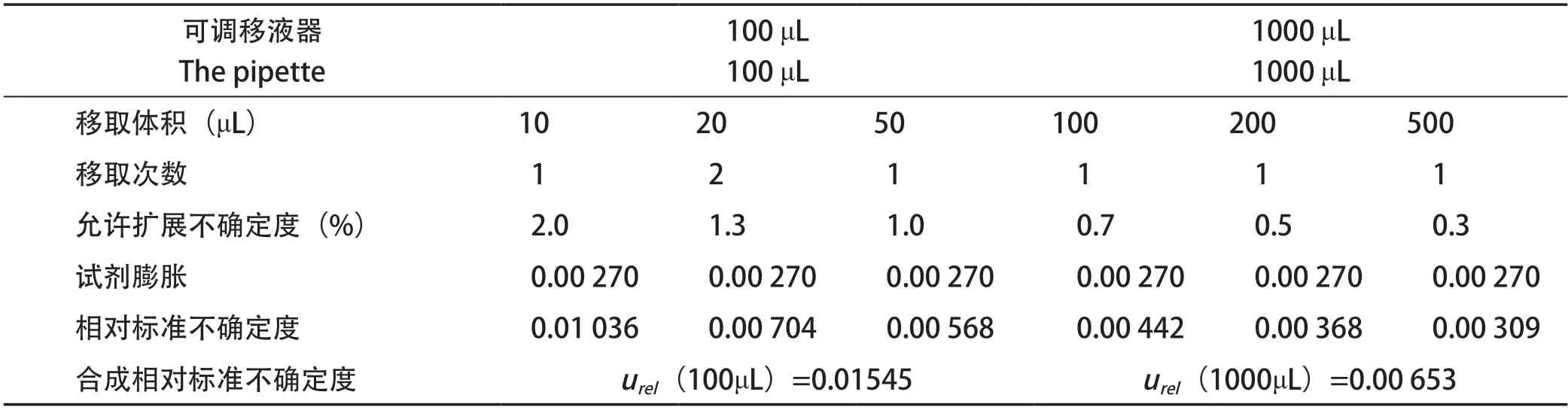
表2 稀释过程中可调移液器引入的不确定度Table 2 Uncertainty of the pipette in the dilution process
综上,标准工作曲线配制引入的相对标准不确定度urel(C3)=0.02379。
2.2.6 标准溶液配制引入的合成不确定度 综上,标准溶液配制引入的相对不确定度为urel(C)
2.3 内标加入体积引入的不确定度 内标配制浓度为100 ng/mL,标准工作曲线定容体积均为10 mL,标准工作曲线中内标加入量为1.0 mL,试样中内标加入量为100 μL,试样经提取定容处理后,标准曲线和试样上机浓度均为10 ng/mL,内标维持在同一水平。内标配制过程产生的不确定度带来的影响可以忽略,只考虑标准曲线中内标加入体积带来的不确定度urel(1000 μL内)和试样中内标加入体积带来的不确定度urel(100 μL内)。内标加入体积引入的不确定度urel(N)计算过程如表3。
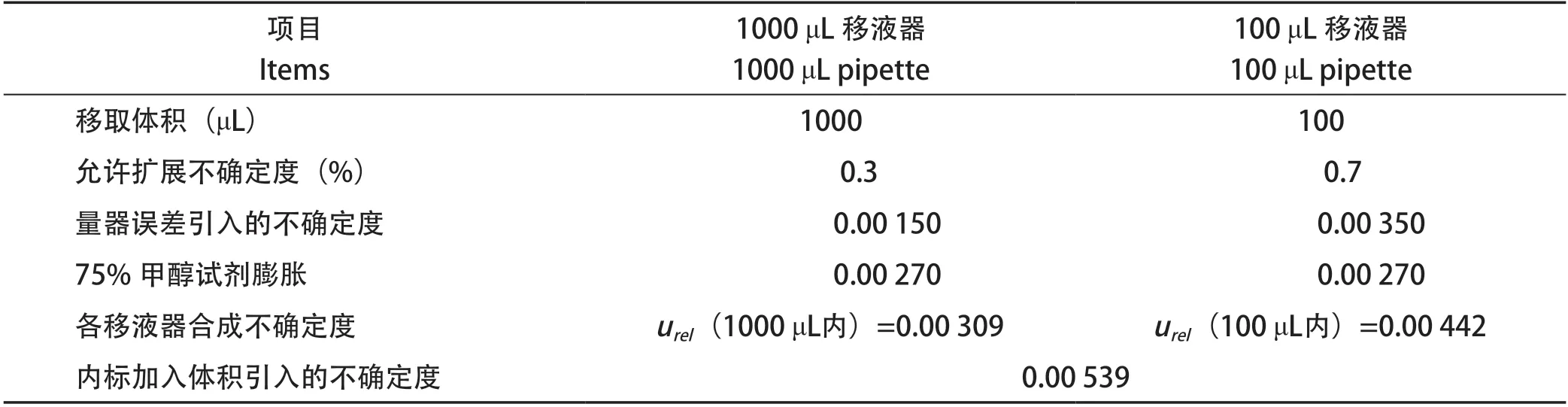
表3 相对标准不确定度urel(N)Table 3 Relative standard uncertainty urel(N)
2.4 样品称量引入的不确定度 试样经高速均质机匀浆,样品的均匀性引入的不确定度可忽略。根据分析天平的校准证书,最大允许偏差为0.01 g,称量样品2.00 g,服从矩形分布,由称量引起的不确定度urel
2.5 样品前处理过程引入的不确定度 定容步骤为加75%甲醇水溶液1.0 mL。75%甲醇在20℃±5℃条件下的体积膨胀系数为0.000 937℃-1,其相对不确定度为0.00 270。1.0 mL使用1000 μL移液器移取,其允许扩展不确定度为0.3%,引入的不确定度为0.00 150。前处理过程引入的合成不确定度为urel(E)0.00 309。
2.6 样品重复测定引入的不确定度 在空白基质中添加DNC标准物质,制成5 μg/kg的阳性样品,重复测定6次,计算样品重复测定引入的不确定度,结果如表4。
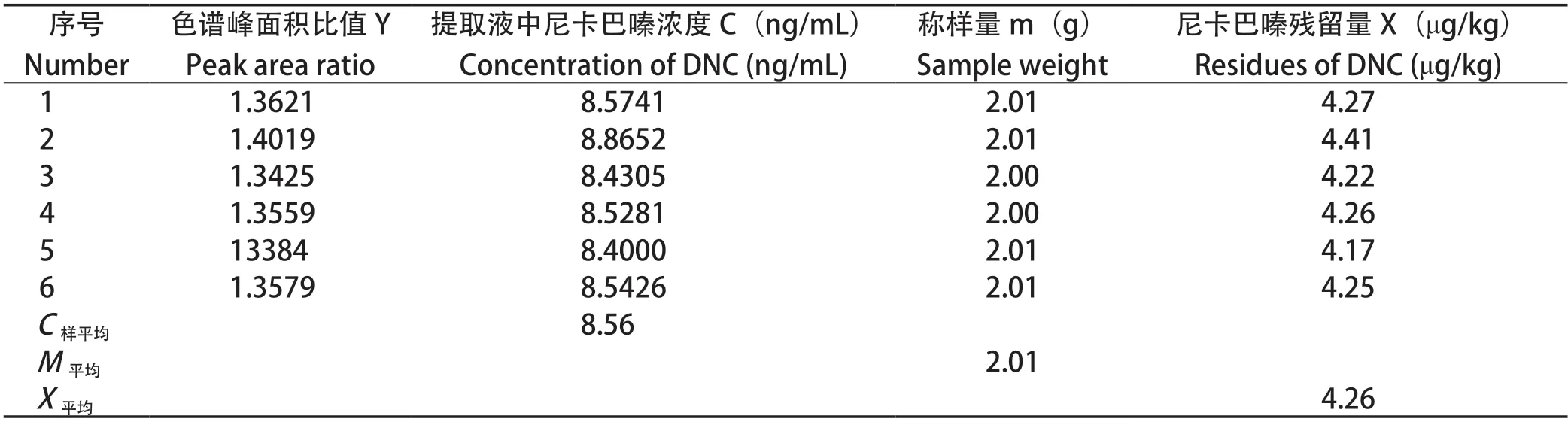
表4 鸡肉中尼卡巴嗪残留标志物含量重复测定结果Table 4 Repeated determination results of nicarbazine residue marker in chicken
样品重复测定引入的不确定度为A类不确定度,根据表5重复测定结果,计算样品重复测定的标准偏差为:=0.08050;样品重复测定的标准不确定度U(X)==0.03286;样品重复测定引入的相对标准不确定度urel(X)==0.00771。

表5 4,4′-二硝基均二苯脲标准曲线测定结果Table 5 Results of 4,4′-Dinitrocarbanilide standard curve
2.7 标准曲线拟合引入的不确定度 系列浓度为2、10、20、50、200、500 ng/mL的标准曲线中含有浓度为10 ng/mL的内标溶液。因使用内标校准,则以待测物峰面积和内标物峰面积的比值为横坐标,以待测物浓度和内标物浓度比值为纵坐标,建立标准曲线,标准曲线测定结果如表5。
其中,p为标准溶液测定次数(p=6);
标准曲线拟合引入的标准不确定度:U(Y)
其中:SR为标准曲线拟合的剩余标准差;a为曲线的斜率;n为样品重复测定次数(n=6);Ci为标准溶液各点的浓度;C样平均为重复测定样品提取液DNC浓度的平均值;C平均为标准溶液各浓度的平均值;p为标准溶液测定次数(p=6)。
2.8 回收率引入的不确定度 在鸡肉中分别添加质量浓度为5、25、50 μg/kg的目标分析物,每个浓度作3个平行,回收率计算结果如表6,回收率引入的相对标准不确定度urel(fR)=0.02411
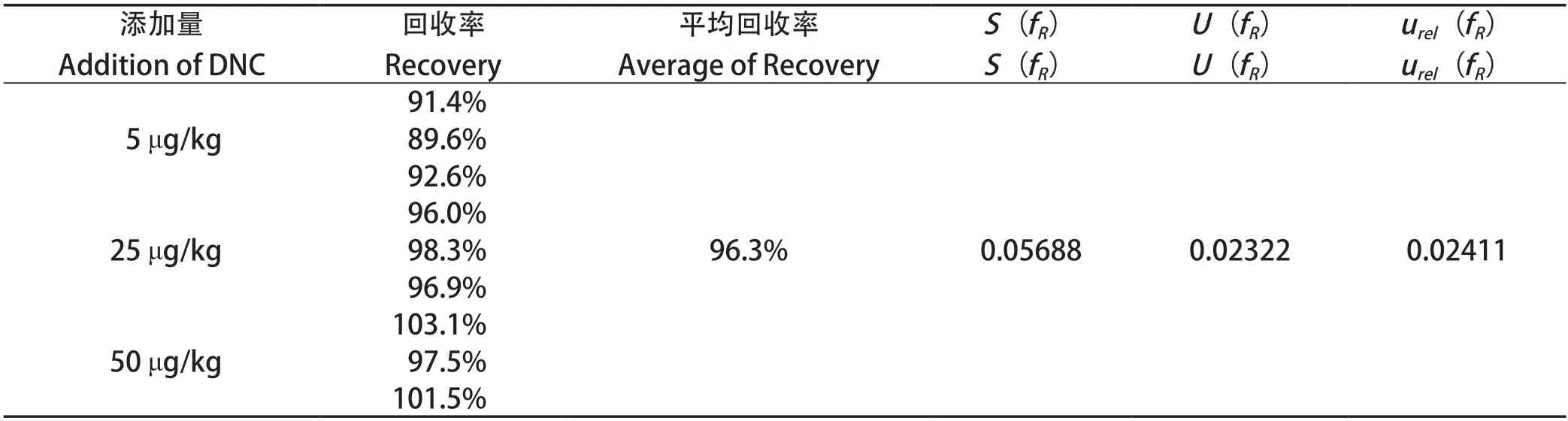
表6 回收率测定结果与不确定度Table6 The recovery and uncertainty of pesticides
2.9 合成标准不确定度及贡献率 根据各分量评定的相对标准不确定度,合成尼卡巴嗪残留标志物残留量的测量相对标准不确定度,并评定各个分量对测量不确定度的贡献率,结果如表7。依据JJF1059.1-2012,不确定度为U(X)=4.26×0.005459=0.23 μg/kg,扩展不确定度为U=K×U(X)=0.46 μg/kg。因此,用液相色谱质谱法测定鸡肉中尼卡巴嗪残留标志物的含量时,扩展不确定为0.46 μg/kg(k=2),试样中尼卡巴嗪残留标志物的含量表示为:4.26±0.46 μg/kg(k=2)。各分量对合成不确定度的影响从大到小依次为标准溶液配制、回收率、标准曲线拟合、样品重复测定、前处理过程、内标加入体积、称量。
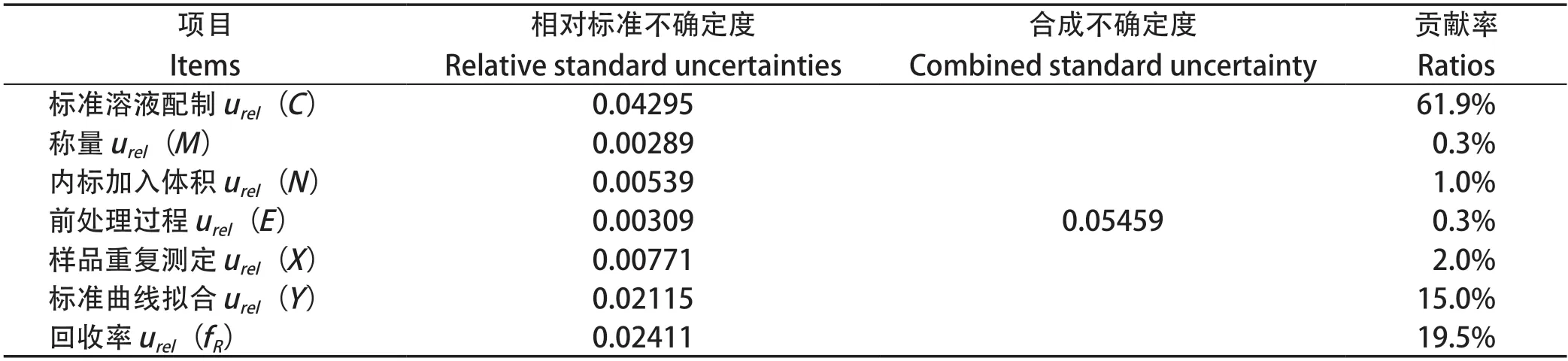
表7 尼卡巴嗪残留标志物残留量测定的合成不确定度Table 7 The relative standard uncertainty of nicarbazine residue marker
3 讨论
本文按照GB 29690-2013对鸡肉中尼卡巴嗪残留标志物残留量进行检测,通过对整个试验过程不确定度来源的分析和评定,确定影响测量结果不确定度的主要因素为标准溶液配制、回收率和标准曲线拟合。评估结果与蒙丽琼等[23]研究基本一致,但三个因素的顺序不同。考量原因,与标准物质自身的不确定度和稀释过程有关。黄荣浪等[24]认为不论试验结果是否折算回收率,回收率对试验结果的不确定度均存在显著影响,与本文评定结论一致。已报道的对回收率的不确定评定均采用一个添加浓度的平行测定,本文对高、中、低三个浓度进行评定,主要考虑了实际待测样品残留量的不确定性。李文杰等[25]评定水产品中氟苯尼考残留量测量不确定度的结论为标准曲线工作液配制、曲线拟合是主要因素,回收率的影响并不突出,考虑可能与所评定目标物本身的性质和检测方法有关。
为完善该检测方法的质量控制体系,在日常工作中应对标准溶液配制、回收率和标准曲线拟合等几个关键步骤严格控制,保证检测结果质量。标准溶液配制过程对合成不确定的贡献率较大,主要与量器误差、量器使用频次,以及实验人员的操作水平有关[26-27]。在配制标准溶液时,应选择国家认可的标准物质中心生产的合格、有效的标准物质,尽量减少稀释定容过程,考虑待测样品实际含量的不确定性和仪器的检测性能,选择合适梯度浓度的标准工作曲线,降低由标准曲线拟合引入的不确定度。建议在进行准确度要求较高的测量时,如:能力验证、盲样考核、检出值在限量值附近等情况时,标准曲线方法得到初步结果后,选择单点校正的方式进行确定,可有效降低检测过程中的不确定度。回收率体现了检测过程中对目标物的提取和净化效率,可通过优化前处理过程、配制基质标液降低机制效应等方法提升加标回收率。此外,试验过程中使用的量器必须检定合格;加强仪器的维护保养,保证测定时仪器处于最佳状态;提高检测人员的操作水平,配合多种质控手段,有效控制和提高实验室的数据质量,为药物残留量测量结果的科学性提供依据。
猜你喜欢
山西文学(2023年6期)2023-06-09
化工管理(2021年7期)2021-05-13
广东茶业(2019年2期)2019-06-18
农药科学与管理(2019年12期)2019-05-20
中成药(2018年1期)2018-02-02
中国农资(2016年1期)2016-12-01
色谱(2015年6期)2015-12-26
化工进展(2015年3期)2015-11-11
中国药业(2014年24期)2014-05-26
无机化学学报(2014年8期)2014-02-28
