
表没食子儿茶素没食子酸酯药动学及其药物相互作用研究进展
2023-08-31 02:29陈思文吕月张凯娜
中国药理学与毒理学杂志 2023年8期
陈思文,吕月,张凯娜
(中国人民解放军总医院第二医学中心1.消化内科,国家老年疾病临床医学研究中心,2.保健三科,北京 100853;3.西安交通大学医学部基础医学院药理学系,陕西西安 710061)
茶多酚是茶叶中多酚类物质的总称,由30余种多酚类物质组成,其中儿茶素占茶多酚总量的60%~80%,而儿茶素中含量最高的几种单体组分分别是表没食子儿茶素没食子酸酯(epigallocatechin gallate,EGCG)、表儿茶素(epicatechin,EC)、表没食子儿茶素(epigallocatechin,EGC)和表儿茶素没食子酸酯(epicatechin gallate,ECG),其中EGCG(图1)是2-连苯酚基苯并吡喃与没食子酸形成的酯,抗氧化能力最强,同时因其结构中有6个邻位酚羟基,许多性质优于其他儿茶素。EGCG 在抗肿瘤、抗感染、防辐射损伤、改善脂代谢、抗氧化等方面均显示出良好的应用前景[1-2]。近年研究表明,EGCG 还能抑制新型冠状病毒繁殖,阻止其进入靶细胞及继发炎症风暴,有望用于防治新型冠状病毒肺炎[3],但要应用于临床,需考虑其药动学特点及与其他药物的相互作用。本文就近年来关于EGCG的药动学特点及其对肝药物代谢影响的研究进展进行综述。

图1 表没食子儿茶素没食子酸酯(EGCG)的结构.
1 EGCG体内药动学特点
1.1 体内吸收和分布
EGCG 的口服生物利用度差。给人类健康志愿者饮用绿茶溶液后,大部分EGCG 不进入血液,而是通过胆汁排至肠道[4]。在男性健康志愿者中,口服6 种不同剂量(50~1600 mg)EGCG 后,血药浓度峰值(maximum concentration,Cmax)为130.37~3391.60 μg·L-1,血浆药物浓度-时间曲线下面积(area under the curve,AUC)为441.66~10367.98 μg·h·L-1,达峰时间(time of maximum concentration,Tmax)约为1.31~2.19 h,而EGCG 163.8 μmol·kg-1ig 给予小鼠后的Cmax为(0.28±0.08)μmol·L-1,Tmax为(90±26)min[5]。Suganuma等[6]ig 给予小鼠3H 标记的EGCG 后,在胃肠、肝、肺、胰腺、乳腺和皮肤中检测到显著的放射性,脑、肾、子宫、卵巢和睾丸中也发现放射性分布;间隔6 h 后,对雌鼠二次给药,其血液、脑、肝、胰腺、膀胱和骨骼中的放射性水平比单次给药提高4~6倍。表明EGCG 口服给药后大部分进入肠道,其代谢产物在体内分布广泛,多次给药会有累积效应。美国药典进行的膳食补充剂安全数据审查结果也显示,禁食期间反复口服大剂量绿茶提取物,可显著提高儿茶素(尤其是EGCG)的生物利用度[7]。
EGCG iv 给药与ig 给药的体内分布不同。大鼠ig 给药后EGCG 主要分布在肠道,肺和肝中分布较少;而iv给药后主要分布在肝、肺和小肠[8]。大鼠iv 给予EGCG 10 mg·kg-1后Cmax,AUC 和Tmax分别为(8.92±2.68)g·mL-1,(161±39)mg·min·L-1和(24±7)min[5]。
1.2 代谢和排泄
1.2.1 EGCG 口服给药后经肠道细菌代谢和经尿、便排泄
EGCG 在口服给药后大部分进入肠道。Kohri等[9]ig 给予给大鼠3H 标记的EGCG 后发现,给药后72 h 内,粪便中的放射性为总放射性的35.2%,且血和尿中检测到的放射性主要来源于EGCG 被肠道细菌降解的产物。Li 等[10]报道,利用人类粪便菌群对EGC,EC 和ECG 进行厌氧发酵的产物与口服摄入EGCG 的人血和尿中鉴定的3 种儿茶素环状裂变产物相同,也说明口服EGCG 被排入肠道后在肠道细菌的作用下进行代谢。动物及人的体内外研究显示,EGCG 在肠道微生物分泌的水解酶、氧化还原酶、裂解酶和转移酶等的作用下先降解为EGC和没食子酸,随后进一步转化[11]。
1.2.2 EGCG的肝代谢和细胞外排
葡萄糖醛酸化、硫酸化、甲基化和环分裂代谢是儿茶素的主要代谢途径;77%的EGCG 以游离形式存在,且容易发生甲基化。EGCG 在肝微粒体中经尿苷三磷酸葡萄糖醛酸基转移酶(UDP-glucuro⁃nosyltransferase,UGT)代谢,主要产物是EGCG-4′-O-葡糖苷酸,人类参与EGCG 代谢的UGT 主要是UGT1A1,UGT1A8和UGT1A9,其中UGT1A8的催化效率最高[12]。EGCG 还可被人、小鼠和大鼠的胞浆硫转移酶(sulfotransferase,SULT)时间和浓度依赖性硫酸化,其中SULT1A1 主要负责肝中的硫酸化,而SULT1A1 和SULT1A3 主要负责肠道中的硫酸化[13-14]。Li 等[15]研究表明,小鼠EGCG 静脉给药后,血和尿中的主要代谢物是葡萄糖醛酸化和硫酸化衍生物。
主动外排会影响许多化合物的生物利用度,降低其细胞内有效浓度。研究表明,EGCG 的外排主要通过多药耐药相关蛋白(multidrug resistanceassociated protein,MRP)完成,参与EGCG 外排的MRP 主要是MRP1 和MRP2;其中MRP2 位于肠、肾和肝细胞的顶面,分别将EGCG 从血液中外排到肠道、尿和胆汁中;MRP1 位于肠、肝细胞的基底外侧,将化合物从细胞外排到肠道中[16]。
2 EGCG 对肝药物代谢酶、转运蛋白和肝药物代谢的影响
肝是药物代谢的主要器官。药物的肝内代谢分为Ⅰ相反应和Ⅱ相反应。参与Ⅰ相反应的代谢酶主要是细胞色素P450(cytochrome P450,CYP450)超家族、乙醛脱氢酶和芳基乙酰胺脱乙酰酶(arylacetamide deacetylase,AADAC)等,参与Ⅱ相反应的代谢酶包括N-乙酰基转移酶(N-acetyl⁃transferase,NAT)、UGT、谷胱甘肽S 转移酶(glu⁃tathioneS-transferase,GST)和SULT 等。此外,肝组织中还有药物转运蛋白参与药物的肝脏摄取和药物代谢产物经胆汁的排泄过程,包括P-糖蛋白(P-glycoprotein,P-gp)、MRP、有机阴离子转运多肽(organic anion transporting polypeptides,OATP)和ABC转运蛋白等。
Matić 等[17]利用ADMET 软件对绿茶儿茶素的体内代谢进行分析。结果表明,EGCG 在体内经过甲基化、葡萄糖醛酸化后,不能穿过细胞膜,因此口服生物利用度低,且EGCG 是P-gp、UGT 同功酶和CYP2C9 的底物,提示EGCG 可能影响肝的药物代谢。上述结论是通过计算机软件分析得出的。目前更常用的研究方法是体外制备微粒体,用已经明确通过该代谢酶代谢的底物作为探针,检测代谢酶的活性,以判断新药对代谢酶活性的影响,或者直接临床观察新药对另一种药物药动学的影响。
2.1 对Ⅰ相代谢酶及相关药物代谢的影响
参与Ⅰ相代谢反应的代谢酶主要是CYP450超家族、乙醛脱氢酶和AADAC 等。CYP450 根据氨基酸序列的同源程度可以分为CYP1A1/2,CYP2B6,CYP2C8,CYP2C9,CYP2D6,CYP2E1 和CYP3A 等亚型。大部分研究显示,EGCG 抑制CYP450 活性。Misaka 等[18]通过体外研究发现,EGCG 对人肝微粒体中CYP2B6(底物为安非他酮)和CYP2C8(底物为阿莫地喹)的活性产生竞争性抑制;对人肝微粒体和人肠道微粒体中CYP3A(底物为咪达唑仑)的活性产生非竞争性抑制。Satoh等[19]用同样方法研究发现,EGCG 抑制人肝微粒体中CYP3A4(底物为酮康唑)和CYP2C9(底物为磺胺苯吡唑)活性;对人肝微粒体中CYP1A2(底物为呋拉茶碱)活性有强竞争性抑制作用,但是对CYP2D6(底物为奎尼丁)活性没有影响。降压药物氨氯地平主要经肝代谢。Han 等[20]研究报道,EGCG 能升高大鼠体内氨氯地平的Cmax和AUC,降低其Tmax,明显延长该药的代谢半衰期,利用大鼠肝微粒体培养系统研究发现,EGCG 的这种效应与其对CYP3A活性的抑制有关。
Ikarashi 等[21]研究认为,由于EGCG 减少小鼠肠道梭菌属细菌,由该细菌产生的石胆酸减少,肝中石胆酸水平降低,导致孕烷X 受体向肝细胞核移位减少,进而导致CYP3A 表达水平降低。Weng等[22]研究则认为,是EGCG 结合了CYP450 的活性位点,或与CYP450 形成了蛋白质聚集体,进而抑制了CYP450 活性;谷胱甘肽能结合体内有毒物质,保护机体免受其伤害,是体内重要的解毒物质,该研究显示EGCG 的上述效应可被谷胱甘肽消除,表明只有当谷胱甘肽因解毒作用被耗尽时,CYP450 的表达或活性才会被EGCG 抑制。也有少数研究不支持EGCG 对CYP450 的抑制作用,如Dudka 等[23]研究报道,EGCG 可增强肝微粒体CYP450活性。
AADAC 参与抗结核药利福平的水解过程,可将利福平水解成无活性的代谢物后经尿液排出,该代谢产物与利福平的肾毒性相关。Yasuda 等[24]在人肝微粒体的研究表明,EGCG能明显抑制AADAC的活性,减少利福平水解,表明EGCG 可能会增强利福平的药效,并减轻利福平的肾毒性。
2.2 对Ⅱ相代谢酶及相关药物代谢的影响
参与Ⅱ相代谢反应的代谢酶包括NAT、UGT、GST和SULT等。
人类NAT 有NAT1 和NAT2 亚型,其中NAT1是5 氨基水杨酸(5 aminosalicylic acid,5-ASA)的乙酰基代谢酶。5-ASA临床用于治疗溃疡性结肠炎,经NAT1 代谢后形成无抗炎作用的N-乙酰-5-ASA。EGCG 非竞争性抑制人胆管癌细胞系KMBC 中的NAT1 活性。提示EGCG 可能会抑制5-ASA 的代谢,增强其疗效[25]。
UGT 在肝中的活性高,已发现其19 个同工酶。Mohamed 等[26]研究报道,EGCG 能抑制人肝微粒体UGT1A4(底物为三氟拉嗪)的活性。三氟拉嗪是一种抗精神病药,经肝代谢产生多种活性代谢产物,临床上需注意EGCG 可能会增加三氟拉嗪的抗精神病疗效;EGCG 能抑制人肝微粒体UGT1A1(底物为β 雌二醇)的活性,提示应关注EGCG 对性激素的影响;EGCG 对人肝微粒体UGT1A9(底物为麦考酚酸)的活性无影响,表明EGCG 不会影响免疫抑制药麦考酚酸的疗效。
SULT 能利用3′-磷酸腺苷5′-磷酸硫酸将磺酸基团转移到含有羟基或氨基的底物化合物中以产生更适合从体内清除的极性产物。SULT1A1 是成人肝中的SULT 亚型之一。Cook 等[27]研究显示,SULT1A1含有2个非相互作用的变构位点,分别结合儿茶素和非甾体类抗炎药,EGCG 对SULT1A1表现出高度的亲和力和特异性,其与SULT1A1 的儿茶素结合位点结合后能控制酶活性位点的开闭,影响相关药物的体内代谢。
2.3 对药物转运蛋白及相关药物代谢的影响
肝中还有药物转运蛋白参与药物的肝摄取和药物代谢产物经胆汁的排泄过程,包括P-gp、MRP、OATP和ABC转运蛋白等。
OATP 在体内分布广泛,其中OATP1B1 主要分布于肝细胞基底膜上,参与许多药物的肝摄取过程,由其介导的药物摄取速率决定了药物总清除率的高低。瑞舒伐他汀(rosuvastatin)和辛伐他汀(simvastatin)是常用的冠心病治疗药物,它们都是OATP 的作用底物,Huang 等[28]研究报道,EGCG明显增加瑞舒伐他汀的AUC;在OATP1B1-HEK293T 细胞模型中,EGCG 可使OATP1B1 介导的瑞舒伐他汀摄取量降低至对照组的45.61%;表明EGCG通过抑制OATP1B1增加瑞舒伐他汀的血药浓度,减少瑞舒伐他汀在肝的分布,可能会降低瑞舒伐他汀的药效。Zeng 等[29-30]先后针对健康男性志愿者进行了2 项开放标签、三阶段随机交叉药动学研究,评估EGCG(800 mg,每天1 次)对瑞舒伐他汀和辛伐他汀药动学的影响。结果显示,EGCG 显著降低瑞舒伐他汀的全身暴露量,但不影响其消除半衰期,这种作用可能会影响瑞舒伐他汀的降脂作用;EGCG 对辛伐他汀药动学总体无影响,但SLCO1B1521TT基因型(OATP1B1 由SLCO1B1基因编码)志愿者辛伐他汀酸的AUC值降低,表明EGCG 与辛伐他丁的相互作用受SLCO1B1基因型相关功能的影响。
Tan 等[31]研究报道,对服用纳多洛尔(nadolol)的雄性自发性高血压大鼠ig给予EGCG(10 mg·kg-1)后,回肠OATP1A5和有机阳离子转运体1 mRNA的表达水平下降,体内纳多洛尔的Cmax和AUC 降低,生物利用度和降压效果下降,心血管事件风险增加。
ABC 转运蛋白位于肝细胞胆管侧膜中,可逆浓度梯度将药物及其代谢物泵出肝细胞到胆汁中。尼达尼布(nintedanib)是治疗特发性肺纤维化的口服小分子激酶抑制剂,是ABCB1 的底物,而EGCG是有效的ABCB1 调节剂。一项针对特发性肺纤维化患者进行的双阶段随机交叉药动学研究显示,服用茶多酚(500 mg,每天2 次,其中EGCG>60%)后7 d 就能使尼达尼布的暴露减少21%,Cmax降低14%,这种效应在ABCB13435C>T 野生型变异的患者中更为明显[32]。
2.4 对其他临床常用药物肝代谢的影响
对乙酰氨基酚(acetaminophen)是西方国家药物性肝损伤最常见的药物之一,其中间代谢产物对肝有毒性作用。研究表明,喂饲EGCG 能通过降低CYP3A,CYP2E1,UGT 和GST 的活性,减少血浆和肝中中间代谢产物的含量,减轻乙酰氨基酚诱导的大鼠肝功能损伤[33](表1)。
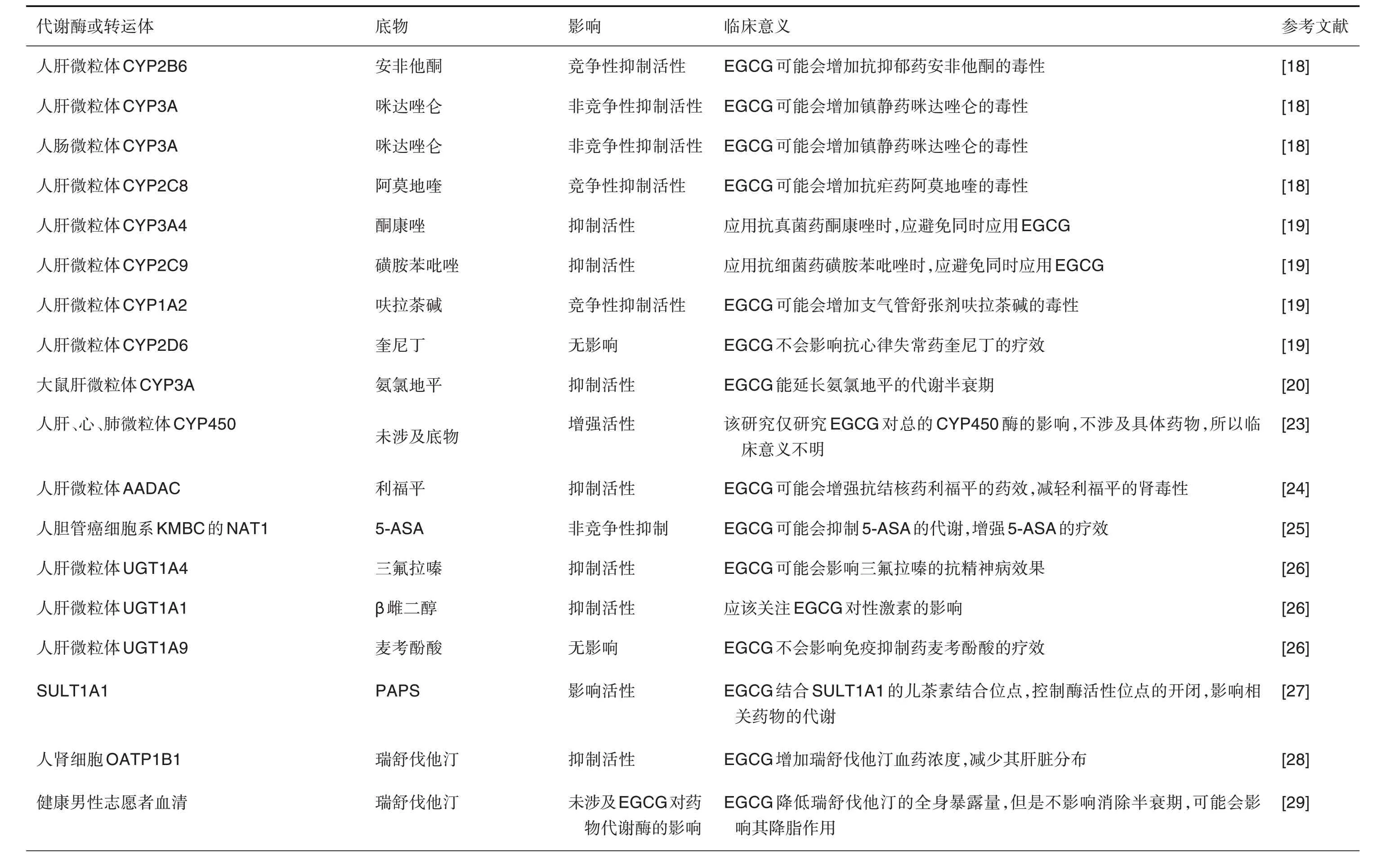
表1 EGCG对药物代谢酶、转运体或药动学的影响及其临床意义
他莫昔芬(taximofen)为合成的抗雌激素药物,临床上用于治疗乳腺癌和卵巢癌,该药主要在肝代谢,代谢产物也有抗雌激素活性。Shin 等[34]在大鼠体内研究发现,EGCG 通过抑制P-gp 和CYP3A 活性,降低其肠道和肝的首过代谢,显著增加他莫昔芬及其代谢物的AUC 和他莫昔芬的Cmax,明显提高他莫昔芬的生物利用度。但Braal 等[35]进行了单中心随机交叉试验评估绿茶胶囊(1.0 g,每天2 次,含300 mg EGCG)对接受他莫昔芬治疗至少3个月的患者的药动学的影响,发现两者之间并无药动学相互作用,认为服用他莫昔芬的患者可同时饮用绿茶(表1)。
比索洛尔(bisoprolol)是临床上常用的降压药物,其不良反应包括引起心率减慢,该药50%经肝代谢。Zeng等[36]研究报道,给自发性高血压大鼠ig给予比索洛尔的同时,ig给予EGCG(100 mg·kg-1)能降低比索洛尔的Cmax,延长其Tmax,减弱其降低心率的作用,增强其降压作用(表1)。
他克莫司(tacrolimus)和环孢素A 均为免疫抑制剂,用于预防同种异体器官或组织移植后的排异反应。Huang 等[37]研究报道,给大鼠iv 或ig 给予他克莫司同时给予EGCG,他克莫司的Cmax和AUC 降低,而表观分布容积和清除率升高;如ig 给予大鼠环孢素A 的同时给予EGCG,低剂量EGCG(3~30 mg)能增加环孢素A 的Cmax和AUC,高剂量EGCG(100 mg)则减少环孢素A 的Cmax和AUC;EGCG 对他克莫司和环孢素A 的药动学的影响与EGCG 抑制药物代谢酶和转运带白(CYP3A1,CYP3A2,UGT1A1 和MRP2)的表达和活性有关(表1)。
3 结语
综上所述,EGCG 是具有临床应用前景的化学物质,但因其口服生物利用度低,限制临床应用。已有多个研究团队尝试用结构修饰和纳米药物传递系统等方法提高EGCG 的稳定性和生物利用度并获得了成功[38-41]。EGCG 对多个肝药物代谢酶和药物转运蛋白的活性有抑制作用,临床上应注意EGCG 和相关药物的相互作用。其中,对于肝代谢产物具有生物活性的药物(如安非他酮、阿莫地喹、呋拉茶碱和三氟拉嗪),EGCG 会增加药物的毒性;而对于经过肝代谢后活性下降、不良反应增加的药物(如利福平、对乙酰氨基酚、5-ASA 和氨氯地平),EGCG 会提高药物的疗效,减轻药物的不良反应;对于因药物转运蛋白作用导致在靶器官浓度降低的药物(如瑞舒伐他汀和尼达尼布),EGCG 也会降低药物疗效。但目前关于EGCG 药动学和药物相互作用的研究结果大都来自于体外细胞或动物研究,不能作为产生人体内作用的直接依据。
猜你喜欢
食品安全导刊(2021年20期)2021-08-30
中成药(2019年12期)2020-01-04
天然产物研究与开发(2018年9期)2018-10-08
中成药(2017年10期)2017-11-16
中成药(2017年5期)2017-06-13
中成药(2017年5期)2017-06-13
中成药(2016年4期)2016-05-17
兽医导刊(2016年12期)2016-05-17
特产研究(2015年1期)2015-04-12
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28
