
CeO2/TiO2吸附剂煤气脱汞产物的浸出规律
2023-08-16 09:07陆洋周劲松周启昕王瑭刘壮李博昊周灵涛
化工进展 2023年7期
陆洋,周劲松,周启昕,王瑭,刘壮,李博昊,周灵涛
(浙江大学能源清洁利用国家重点实验室,浙江 杭州 310027)
煤气化作为一种高效、清洁的煤炭综合利用技术,近年来受到广泛的关注[1]。但是在煤气化过程中,煤中汞(Hg)会被释放到气相中,煤气中的Hg大部分以单质汞(Hg0)的形式存在,因其具有高挥发性、低水溶性等特性而难以被去除[2-3]。除此之外,煤气中还原性气氛(CO、H2)会抑制Hg0氧化[4]。金属氧化物吸附剂可以通过非均相反应催化氧化Hg0,将其固定于金属氧化物表面,故受到广泛关注。同时,金属氧化物吸附剂能与煤气中H2S、HCl 产生协同效果:H2S、HCl 在吸附表面发生解离,形成促进Hg0催化氧化的活性物质(S*、Cl*),可与Hg 结合在吸附剂表面形成HgCl2、Hg2Cl2、HgS和HgSO4等含汞化合物[5-6]。
金属氧化物吸附剂的后续处理是目前亟需解决的难点之一。通常含汞金属氧化物吸附剂处置方式为直接掩埋,但其长时间暴露在自然环境中,受到细菌、酸液等外界环境因素的作用,脱汞产物中的汞可能再次被释放,再次进入大气、土壤、水源中,造成二次污染[7-10]。尤其是吸附剂表面中的HgCl2、HgO 等汞化合物在生态环境中易发生甲基化,进入生物链危害人体生命健康。若在对含汞金属氧化物吸附剂进行填埋之前,将含汞物质与之分离,可有效防止汞的二次污染,此外不含汞的吸附剂也便于后续的再生利用,从而实现吸附剂的绿色化、无害化处理。目前常采用的脱附方式是热脱附,其优点是脱附效率高,并且若加入O2可以起到一定的吸附剂再生效果[11-13]。但是热脱附过程中含汞金属氧化物吸附剂中吸附态汞会分解为Hg0重新进入气相中,仍需采用其他的方式捕获Hg0,会带来额外的成本。HgO、α-HgS(朱砂)分解温度在300℃以上,HgSO4更是在500℃以上才开始分解[14-15]。为使得汞及其化合物尽数与吸附剂分离,热脱附通常设定的温度在500℃以上,故而耗能高也是热脱附的不足之一。
此外,可采用溶液浸出的方式解决金属氧化物吸附剂上脱汞产物后续处理问题。溶液浸出法是指利用液相中的化学试剂使含汞吸附剂表面的脱汞产物迁移至液相中,从而实现吸附剂上汞的脱除。浸出法相较于热脱附,具有温度低、耗能少等优势,后续可添加硫化剂处理含汞溶液[16-17],最终实现金属氧化物吸附剂上吸附态汞的无害化、绿色化处置。Diao等[18]先后使用去离子水、NH4Cl溶液、HCl溶液、HNO3溶液等洗涤含汞废物,发现HgCl2、HgO易被浸出,而HgS难以被溶解。鉴于常见金属氧化物在酸性溶液中会溶解,为避免吸附剂有效组分流失,因此拟采用弱碱性溶液。Issaro 等[19]曾用硫代硫酸钠(Na2S2O3)溶液作为试剂从土壤中浸取汞,结果表明,Na2S2O3能够从土壤有机质中去除与硫基结合的汞。Han 等[20-21]发现在添加Cu2+后形成的铜硫代硫酸盐可与HgS发生反应,从而使得HgS 溶解。S2O2-3会发生水解,使其溶液呈现弱碱性,且其与金属离子有很强的配合能力,可以与Hg2+形成稳定的配合物[22-23]。此外S2O2-3易分解,可利用紫外线分解S2O2-3使得液相中的Hg2+变为HgS从而沉淀[24]。因此本文拟采用Na2S2O3作为溶液的主要成分,浸出金属氧化物吸附剂表面脱汞产物,实现汞的无害化处理。
本文作者课题组先前的研究表明,铈(Ce)基金属氧化物吸附剂在煤气气氛中展现出高效的脱汞能力[3,25-26,28-29]。因此本文使用CeO2/TiO2吸附剂作为研究对象,随后在Na2S2O3溶液中浸出。本文旨在探索一种湿法处置金属氧化物表面脱汞产物的新思路,考察Na2S2O3溶液对于含汞Ce 基吸附剂的脱汞产物浸出效果,探究不同赋存形态的吸附态汞在Na2S2O3溶液中的迁移规律,测试了浸出后对Ce 基脱汞吸附剂吸附效率的影响,为开发金属氧化物脱汞吸附剂湿法再生技术提供基础。
1 实验部分
1.1 CeTi吸附剂制备
选用浸渍法制备Ce 基金属氧化物吸附剂。使用Ce(NO)3·6H2O(AR)作为活性组分,纳米TiO2作为载体,控制CeO2/TiO2的质量比为2∶8[26]。将Ce(NO)3·6H2O与纳米TiO2在去离子水中充分搅拌混合30min后抽滤并干燥12h,随后在500℃、空气中煅烧2h,将吸附剂标记为CeTi。最后将煅烧后的吸附剂研磨过筛,控制CeTi粒径为40~60目。
1.2 汞负载实验
Hg0负载实验在固定床反应装置中进行(图1)。模拟煤气由N2(平衡气和载汞气)、HCl、H2S、NH3、CO、H2组成,气体总流量为1.2L/min。固定床反应器中设有加热丝,在吸附剂位置设有热电偶与温度控制装置相连,可以稳定控制固定床反应装置温度。管路中Hg0的在线浓度检测由RA-915+汞分析单元(Lumex,Russia)完成,最后的尾气处理单元吸收管路中残余的Hg0以及其他有害气体。
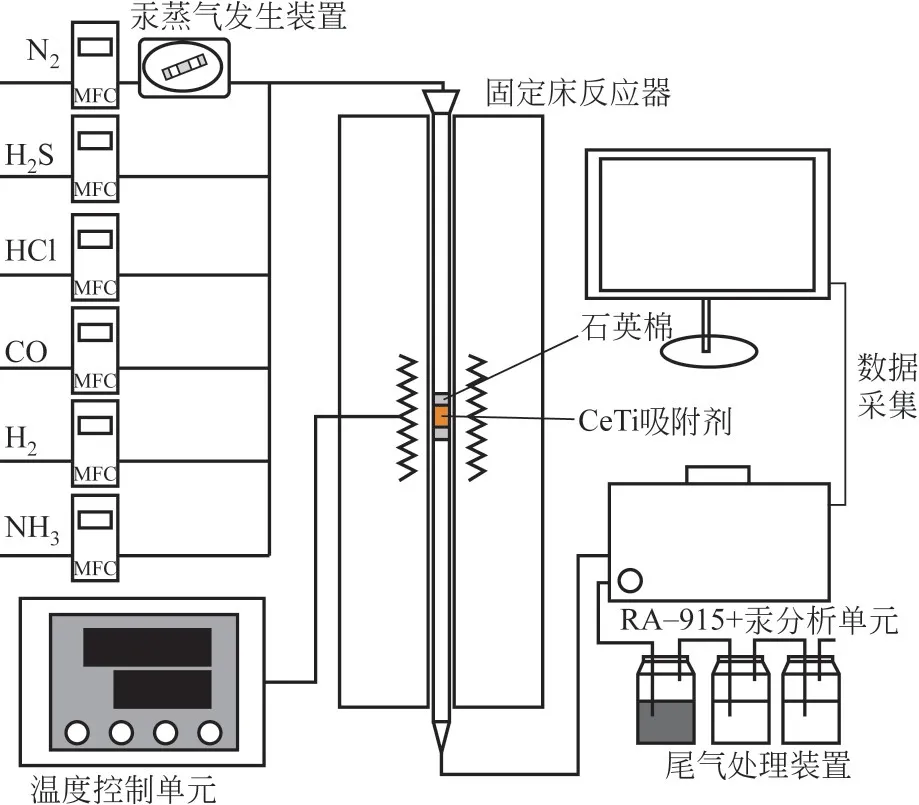
图1 实验装置示意图
为在吸附剂表面负载更多汞以便于后续研究,将吸附时间设定为12h,初始汞浓度控制为80μg/m3,吸附剂用量为0.4g。气相吸附实验气氛、温度以及含汞吸附剂(Hg-CeTi)命名如表1所示。
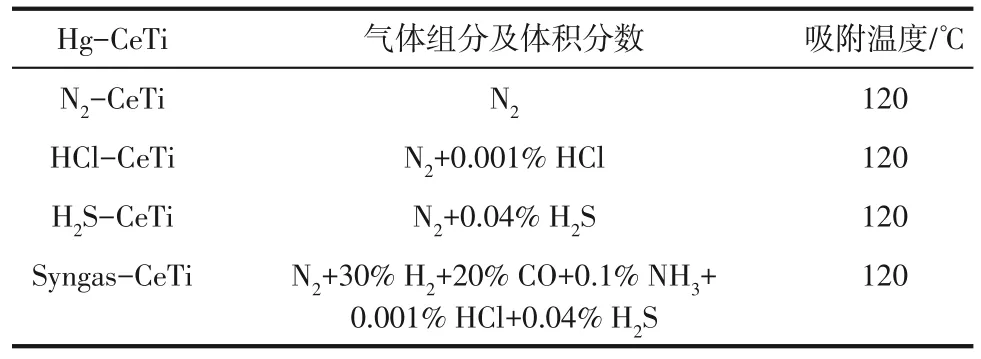
表1 吸附实验气氛及温度
汞程序升温脱附实验(mercury temperature program desorption,Hg-TPD)用于确定Hg-CeTi中脱汞产物中的汞赋存形态,同样在上述实验装置中进行(图1)。鉴于HgSO4的分解温度可达500℃以上,因此设定Hg-TPD 升温范围为20~600℃,升温速率为10℃/min,Hg-TPD 实验中Hg-CeTi 用量为20mg。为排除气体对热脱附的影响,在Hg-TPD实验中仅使用N2,流量为0.5L/min。对Hg-TPD 曲线进行分峰拟合,并统计各个脱附峰的面积比例,即可获得Hg-CeTi表面各类汞化合物的占比。
1.3 吸附汞浸出实验
实验中使用Na2S2O3·5H2O(AR)配置Na2S2O3溶液,为研究不同浓度的影响,分别配置0.1mol/L、0.2mol/L、0.5mol/L、1.0mol/L 和2.0mol/L 五种不同浓度的Na2S2O3溶液。浸出实验中,Hg-CeTi质量与Na2S2O3溶液体积的比例为50mg∶15mL,反应时间为1h,并用磁力搅拌器进行搅拌。汞浸出过程全程采用密封菌种瓶作为反应容器以减少汞的挥发。Na2S2O3溶液浸出后Hg-CeTi 分别命名方式为x-ATMO-CeTi,x代表浓度为xmol/L,ATMO 代表实验气氛,例如0.1-Syngas-CeTi 是指Syngas-CeTi在0.1mol/L Na2S2O3溶液中浸出后所得样品。利用RA-915+取样检测汞元素含量,折算后获得50mg Hg-CeTi中汞元素质量,记为m,单位μg。
汞浸出率(η)计算如式(1)所示。
式中,m0为浸出前Hg-CeTi 所含汞元素质量;mx为xmol/L Na2S2O3溶液浸出后样品所含汞元素质量。
1.4 汞吸附效率测试
汞吸附效率测试在图1的装置中进行,吸附剂用量为100mg,将汞浓度降低为50μg/m3,气体组分、流量以及吸附温度与Syngas-CeTi 组一致,吸附时间为2h。汞吸附效率定义式如式(2)所示。
式中,Cin和Cout分别为固定床反应器进出口单质汞浓度。
2 结果与讨论
2.1 N2-CeTi吸附态汞浸出
HgO为金属氧化物吸附剂在煤气中常见的吸附产物,气相中的Hg0通过物理吸附于CeTi表面,随后部分吸附态Hg0被吸附剂中的活性氧(O*)氧化形成HgO[29]。图2(a)中185℃处的峰对应Hg0的热脱附峰,300℃处对应HgO[30],,此结果与先前研究的吸附机理相符。
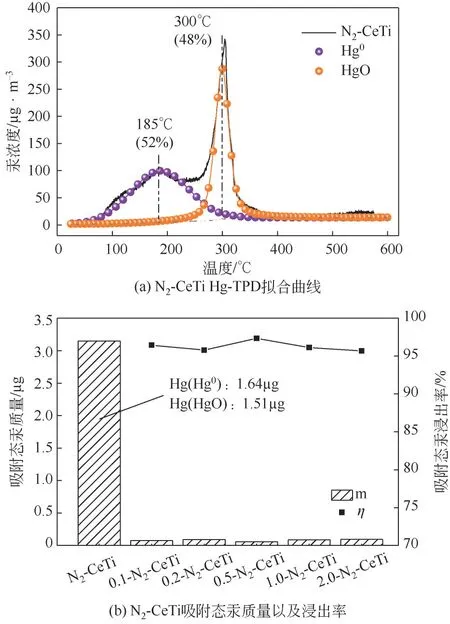
图2 N2-CeTi表面脱汞产物形态以及浸出率
图2(b)所示,N2-CeTi 在不同浓度的Na2S2O3溶液中η都在95%以上,因此推测HgO 易迁移至Na2S2O3溶液中。同时,提高Na2S2O3溶液浓度,汞浸出率出现波动,并未出现明显增强浸出的效果,可以认为0.1mol/L Na2S2O3溶液足以使得HgO 从CeTi 表面迁移至液相。这与Han 等[21]的研究相符,HgO 可以与S2O2-3形成可溶性配合物,反应式为式(3)。
2.2 HCl-CeTi吸附态汞浸出
HCl 是煤气中一种对Hg0在吸附剂表面非均相氧化起显著作用的成分。如图3(a)所示,HCl-CeTi表面吸附态汞大部分以HgCl2的形式存在,此形态的汞元素占总汞的76%,其他以HgO 的形式存在,并且未检测到明显的Hg0峰。这是因为HCl 在CeTi表面会形成活性氯(Cl*),随后吸附态的Hg0会与Cl*结合形成HgCl2[31]。在Cl*的生成过程中会消耗CeTi 中的O*,因而形成更多HgCl2而非HgO[32]。综上所述,HgCl2是HCl和Hg0在CeTi表面上非均相氧化的主要脱汞产物,且属于易溶性汞化合物[33],在自然环境中易被转化为甲基汞,故其在Na2S2O3溶液中浸出规律值得研究。
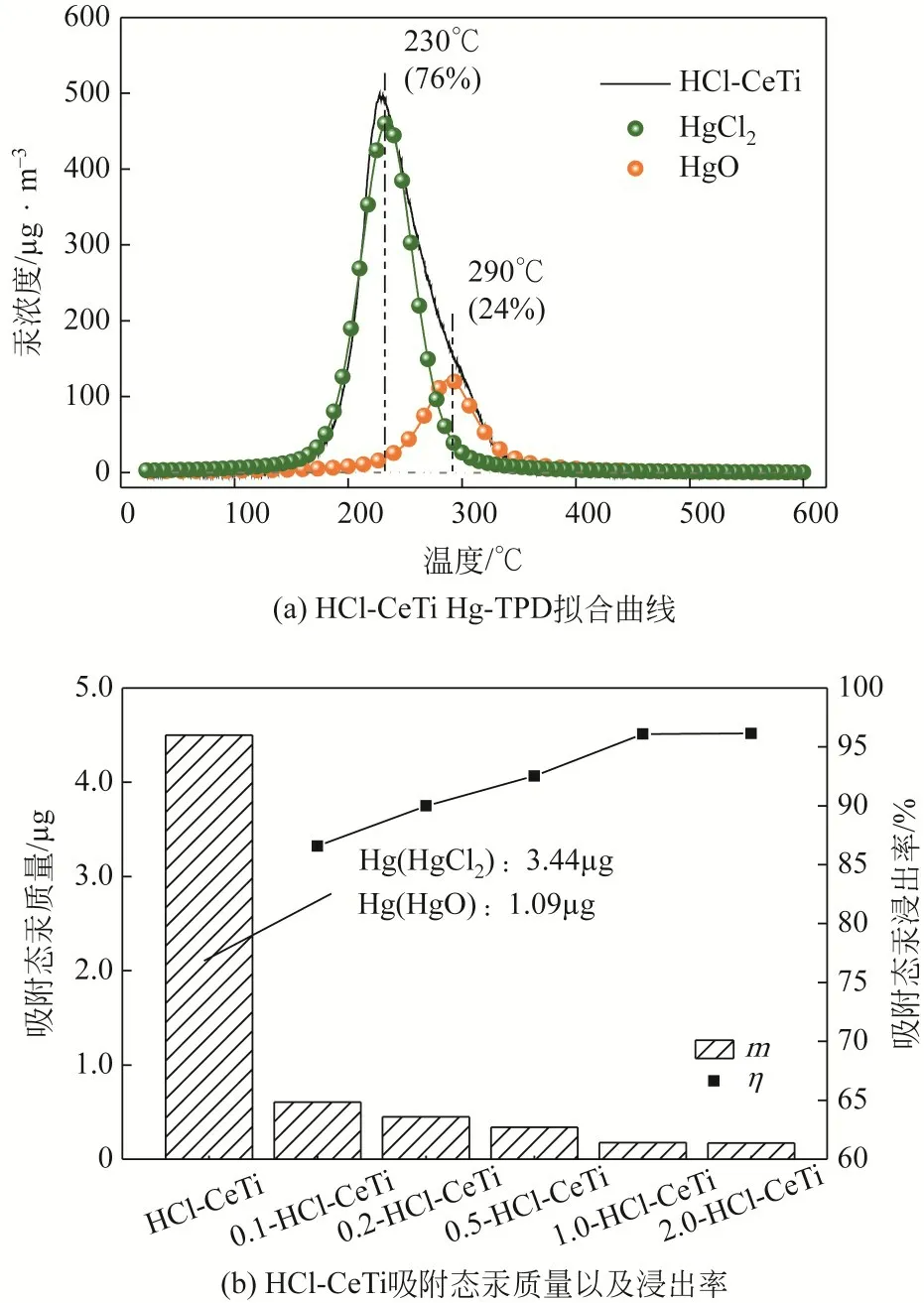
图3 HCl-CeTi表面脱汞产物形态以及浸出率
HCl-CeTi 在Na2S2O3溶液中的浸出效果如图3(b)所示。HCl-CeTi 汞浸出率随着Na2S2O3浓度升高先增加后趋于稳定。在0.1mol/L的Na2S2O3中,η为86.6%,在1.0mol/L和2.0mol/L的Na2S2O3溶液中,η分别上升至96.1%、96.1%。根据前文所述,HgO易溶于Na2S2O3溶液,同时HgCl2属于可溶性汞化合物,然而经0.1mol/L以及0.2mol/L Na2S2O3溶液浸出后,汞浸出率并不如预期,未超过90%。
随 后 利 用Hg-TPD 确 定0.1-HCl-CeTi、0.2-HCl-CeTi表面汞的赋存形态,如图4所示。结果显示,0.1-HCl-CeTi 与0.2-HCl-CeTi 表面仅存在HgO。该结果可能HCl-CeTi 表面吸附态汞在迁移过程中存在转化和竞争所导致。Na2S2O3属于强碱弱酸盐,会发生水解因而其水溶液显碱性,如式(4)[34]。HgCl2属于强酸弱碱盐,在碱性溶液中生成HgO,如式(5)所示[35]。由于OH-被消耗,反应式(4)进一步向右进行,同时S2O2-3的减少会抑制HgO 与S2O2-3的配合反应。故导致部分HgO 在低浓度Na2S2O3中未被浸出。HgCl2向液相中迁移规律可解释为:HgCl2先溶解迁移至Na2S2O3溶液,部分与OH-反应形成HgO,最终HgO 再与S2O2-3反应迁移至液相。

图4 HCl-CeTi浸出后表面吸附态汞赋存形态
2.3 H2S-CeTi吸附态汞浸出
黑色硫化汞[HgS(black)]的分解峰在260℃±15℃[36],图5(a)中255℃的脱附峰对应物质为HgS(black)。红色氧化汞[HgS(red)]的脱附峰温度与氧化汞相近[2,4,15],故图5(a)中290℃的脱附峰可能为HgS(red)与HgO 两种物质的混合脱附峰。此外,并未发现HgSO4的脱附峰。在气相吸附时,H2S 先在CeO2表面与晶格氧O*反应生成活性硫(S*),随后S*直接与气相中的Hg0形成HgS[26]。HgS 作为H2S 和Hg0在CeTi 吸附剂表面的非均相催化氧化产物,是一种十分稳定的汞化合物形态,其浓度积常数(Ksp)仅为2×10-52.7[37]。图5(b)显示,H2S-CeTi的汞浸出率低于N2-CeTi 和HCl-CeTi,Na2S2O3浓度为2.0mol/L时仅为71.9%。此外,H2S-CeTi 的汞浸出率随着Na2S2O3溶液浓度有明显的增加。

图5 H2S-CeTi表面脱汞产物形态以及浸出率
随后,为进一步研究两者不同晶型的HgS以及HgO 共存情况下的浸出规律,分别对x-H2S-CeTi(x=0.1、0.2、0.5、1.0和2.0)中汞赋存形态进行分析,Hg-TPD 结果如图6 所示。图6(a)显示在Na2S2O3浸出前后热脱附峰温度以及区间并未发生明显变化。图6(b)~(f)显示x-H2S-CeTi Hg-TPD曲线的拟合峰在250℃以及300℃附近,为作区分,分别将两个峰对应的吸附态汞记为Hg250和Hg300。结合前文可知,Hg250为HgS(black),而Hg300为HgS(red)和HgO。

图6 x-H2S-CeTi Hg-TPD拟合曲线(x=0.1、0.2、0.5、1.0和2.0)
对图6中Hg250和Hg300的进行统计分析,汇总于图7 中。图7(a)显示,随着Na2S2O3浓度的增加,Hg250的η显著增加,即Na2S2O3可促进HgS(black)的溶解;而Hg300不同于Hg250,η仅轻微增加。结合对于N2-CeTi的浸出规律研究,Hg300中的HgO易迁移至液相,因此在0.1mol/L Na2S2O3溶液浸出后,绝大部分HgO 已迁移至液相,故x-H2S-CeTi 的Hg300应为HgS(red)。结合以上分析,可以推测HgS(red)难以被Na2S2O3溶液浸出。图7(b)中不同形态汞的分布也印证了该结论:由于Na2S2O3溶液对于HgO 的浸出能力强于HgS(black),Hg300相对含量迅速减少;而后Hg300相对含量开始不断增加,表明在Na2S2O3溶液中HgS(red)相较于HgS(black)更难迁移至液相中。Hong 等[38]的研究表明,汞化合物在吸附剂表面稳定性与热脱附峰温度成正比,HgS(red)比HgS(black)的热脱附峰温度更高,与CeTi结合更紧密因此更稳定。因此推测两种不同晶型的HgS向液相迁移能力差异与二者在吸附剂表面的稳定性有关。

图7 x-H2S-CeTi中不同形态汞的质量、浸出率以及分布(x=0.1、0.2、0.5、1.0和2.0)
2.4 Syngas-CeTi吸附态汞浸出
煤气中同时存在多种气体组分(H2、CO、N2、HCl、H2S、NH3等),因而在吸附剂表面吸附态的赋存形态也更为复杂。图8 显示,Syngas-CeTi 表面吸附态汞以HgO、HgCl2和HgS 存在,相对含量分别为45.8%、22.7%和31.5%。对Syngas-CeTi 吸附态汞浸出结果显示,0.1~2.0mol/L Na2S2O3溶液均可使90%以上的汞从Syngas-CeTi 表面脱离迁移至液相,1.0mol/L Na2S2O3溶液中浸出率最高,达到97.3%。

图8 Syngas-CeTi 表面脱汞产物形态以及浸出率
x-Syngas-CeTi的Hg-TPD分析显示,未被浸出的汞以HgS 形态存在。图9(a)为x-Syngas-CeTi 的Hg-TPD曲线对比图,可以发现x-Syngas-CeTi热脱附峰的位置在210℃以及320℃附近,对应浸出前HgS(black)以及HgS(red)的峰位置。其中0.1-Syngas-CeTi左边的峰偏向于230℃,可能是有部分HgCl2还未脱除。在模拟煤气吸附过程中,酸性气体(H2S)会吸附于CeTi表面[6],在低浓度Na2S2O3溶液浸出过程中降低了液相pH,从而抑制HgCl2向HgO 转化,因此并未观察到明显的HgO 峰。除此之外,随着Na2S2O3溶液浓度的增加,210℃峰逐渐减小,而320℃峰几乎不发生变化。在1.0-Syngas-CeTi 仅存在HgS(black)和HgS(red),且316℃的HgS(red)为汞主要赋存形态。综上所述,Syngas-CeTi在汞浸出过程中,表面多种赋存形态的汞向液相迁移难易程度排序为:HgS(red)>HgS(black)>HgCl2>HgO。

图9 x-Syngas-CeTi Hg-TPD曲线对比以及1.0-Syngas-CeTi表面汞赋存形态
2.5 吸附剂浸出后吸附效率
分 别 测 试 了 CeTi、Syngas-CeTi 以 及1.0-Syngas-CeTi 在2h 内的汞吸附效率,结果如图10 所示,三者在2h 内的平均汞吸附效率分别为79.23%、25.15%以及74.93%。Syngas-CeTi 的平均吸附效率已经小于30%,可认为其已处于失活状态。经过溶液浸出后,1.0-Syngas-CeTi 恢复至74.93%,但小于新鲜吸附剂CeTi 的吸附效率。该现象有如下两方面原因:其一,在负载过程中,汞占据活性位点导致Syngas-CeTi 的失活,而1.0-Syngas-CeTi 已浸出表面大部分的汞,因此吸附性能得以恢复;其二,可能由于在浸出过程中无法补充缺失的活性氧,因此1.0-Syngas-CeTi 吸附性能不及CeTi。故溶液浸出法有作为金属氧化物吸附剂再生方式的潜力,但仍需进一步研究。
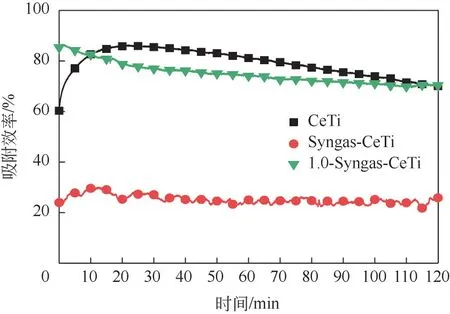
图10 吸附剂汞吸附效率测试
3 结论
研究了煤气中CeO2/TiO2表面脱汞产物及其在Na2S2O3溶液中的浸出行为。在吸附过程中,气相中Hg0在CeTi形成HgO、HgCl2和HgS。在随后的溶液浸出中,HgO易与S2O2-3反应形成可溶性配合物;HgCl2自身属于可溶性汞化合物,但在碱性溶液中会转化为HgO;由于HgS的稳定性,HgS的浸出率低于HgCl2与HgO;HgS(red)较HgS(black)与CeTi 结合更稳定,HgS(red)几乎难以迁移至液相。此外Na2S2O3溶液浓度增加可提高脱汞产物浸出率,促进HgO、HgCl2以HgS(black)向液相迁移,但对于HgS(red)收效甚微。最终Na2S2O3溶液去除了Syngas-CeTi 表面97.3%的脱汞产物,且残余为主要为HgS(red)。结果表明,通过Na2S2O3溶液浸出的方式一方面可为之后吸附剂再生提供便利;另一方面仅有HgS的存在可显著减少后续吸附剂处理中汞二次释放的风险。最后,Syngas-CeTi 经过溶液浸出后,吸附效率从25.15%恢复至74.93%,因此采用溶液浸出的方式有一定的再生效果。
猜你喜欢
化工管理(2022年13期)2022-12-02
能源工程(2021年1期)2021-04-13
陶瓷学报(2020年6期)2021-01-26
自我保健(2020年11期)2020-12-04
中学生数理化·中考版(2018年11期)2019-01-31
教学考试(高考化学)(2018年5期)2018-12-06
中国氯碱(2017年11期)2017-12-06
中国氯碱(2014年7期)2014-08-15
食品工业科技(2014年15期)2014-03-11
河南科技(2014年16期)2014-02-27
