
基于光热效应和pH响应性的聚多巴胺膜靶向纳米粒子的制备及性能评价
2023-08-14 01:39张翔珂高萌萌陈铭董雨萌苏慧阎雪莹
药学研究 2023年7期
张翔珂,高萌萌,陈铭,董雨萌,苏慧,阎雪莹
(黑龙江中医药大学药学院,黑龙江 哈尔滨 150040)
化疗是治疗乳腺癌各个时期的重要治疗手段,为提高单一化疗药物的疗效,目前临床常将两种或多种化疗药物联合使用[1]。除此之外,还可将化疗与其他疗法相结合,例如光疗、免疫治疗等[2-3],以提高对于肿瘤细胞的杀伤能力[4]。介孔二氧化硅纳米粒(mesoporous silica nanoparticles,MSNs)作为近些年来发展起来的一种新型无机介孔纳米材料[8],具有比表面积大以及良好的生物相容性等优点,可在一定程度上增加药物的负载量。尽管MSNs具有多方面的优点,但是载药MSNs存在一定的药物突释风险,为此通常需要在MSNs表面上进行修饰,以实现对MSNs孔道的封堵,控制药物释放。
聚多巴胺(polydopamine,PDA)是盐酸多巴胺在碱性条件下氧化而形成的具有强大黏附性的仿生聚合物[9],可以实现对MSNs表面覆盖,而在肿瘤部位的酸性环境中产生裂解,可防止MSNs孔道内药物在非肿瘤部位的突释。PDA作为光热转化材料在近红外光区有强吸收性[10],经808 nm近红外激光照射后可产生热能,当温度达到一定限度时,对肿瘤细胞产生杀伤作用,可实现化疗和光热治疗的联合作用[11]。另外由于其表面存在着丰富的官能团易实现靶头的连接,可实现纳米粒的主动靶向作用[12]。叶酸受体(folate receptors,FR)作为常用的肿瘤治疗的受体,在正常组织中的表达处于高度保守状态,而作为FR亚型之一的FR-α则在乳腺癌等上皮谱系肿瘤中过度表达[13-14],因此FR-α可能成为乳腺癌的潜在治疗靶点,因此本研究选择叶酸作为纳米粒的靶头。
为构建高效、低毒、高肿瘤靶向性的给药系统,本实验拟制备本实验中选择临床常见配伍方式蒽环类药物和微管抑制类药物中的代表药物盐酸吡柔比星(pirarubicin hydrochloride,THP)[5]和多西紫杉醇(docetaxel,DTX)[6-7]作为模型药,以FR-α为靶点,以包覆聚多巴胺的介孔二氧化硅为载体,将化疗与光疗相结合的纳米制剂对叶酸过表达型乳腺癌进行治疗。本试验中利用单因素考察对FA-PDA-THP-DTX-MSNs制备工艺进行优化,并对其形貌、光热效应、体外释放、体外药效学以及大鼠体内药代动力学进行了考察。从药剂学的角度为实现化疗与光疗相结合的靶向治疗方法提供了新的策略和依据。
1 材料
1.1 试剂与药品多西紫杉醇(批号:C12161879,上海麦克林生化科技有限公司);盐酸吡柔比星[批号:A2110358,凯梅根(上海)生物科技有限公司];盐酸多巴胺(批号:C11956740,上海麦克林生化科技有限公司);三羟甲基氨基甲烷(批号:2021/03,上海麦克林生化科技有限公司);叶酸(批号:0820171004,上海麦克林生化科技有限公司);N-羟基琥珀酰亚胺(批号:C12913790,上海麦克林生化科技有限公司);聚乙烯亚胺(批号:C12417098,上海麦克林生化科技有限公司);甲醇(色谱纯,美国迪马公司);乙腈(色谱纯,美国迪马公司);1-(3-二甲氨基丙基)-3乙基碳二甲胺(上海麦克林生化科技有限公司)。所有购买的试剂均未经进一步纯化直接使用,超纯水由水纯化系统(PALL Cascade III)制备。
1.2 仪器HZS-HA恒温水浴振荡器(哈尔滨市东联电子技术开发有限公司);e2695高效液相色谱仪(美国Waters公司);LR-MFJ-808nm近红外激光器(长春镭锐光电科技有限公司);Nano-ZS90纳米粒度及Zeta电位分析仪(马尔文仪器有限公司);热电偶双通道测温器(东莞市希玛仪表有限公司);ASAP 2460全自动比表面及孔隙度分析仪(美国Micromeritics公司);粉末X射线衍射仪(德国布鲁克公司);STA 449 F5同步热分析仪(德国耐驰公司);FEI Tecnai F30透射电子显微镜(美国FEI公司)。
2 试验方法与结果
2.1 MSNs的制备MSN按照改进的Stöber方法合成[15]并以酸化乙醇回流萃取法去除模板剂。具体操作如下:90 μL三乙醇胺(TEA)与60 mL H2O混合成稀的强碱溶液(pH=9.5),准确称量0.50 g 十六烷基三甲基溴化铵(CTAB)加入溶液中,95 ℃下磁力搅拌30 min后缓慢滴加614 μL 正硅酸乙酯(TEOS),在剧烈搅拌和恒温下反应3 h,离心(8 000 r·min-1,10 min)收集沉淀并用蒸馏水-乙醇交替洗3次,冷冻干燥得到MSN@CTAB白色粉末。
2.2 分析方法建立
2.2.1 色谱条件DTX色谱条件:色谱柱:Kromasil(C18,4.6 mm×150 mm,5 μm);流动相:甲醇:乙腈:水(35∶40∶25,V/V/V);检测波长:230 nm;柱温:25 ℃;流速:1 mL·min-1;进样量:20 μL。
THP色谱条件:色谱柱:Kromasil(C18,4.6 mm×150 mm,5 μm);流动相:乙腈:0.1 mol·L-1醋酸铵溶液醋酸调至pH=4(35∶65,V/V);检测波长:254 nm;柱温:25 ℃;流速:0.8 mL·min-1;进样量:20 μL。
2.2.2 专属性试验按“2.2.1”项下DTX和THP的色谱条件,分别进样DTX和THP对照品溶液、未载药纳米粒(FA-PDA-MSNs)超声上清溶液及载药纳米粒(FA-PDA-THP-DTX-MSNs)超声上清溶液,检测方法的专属性。检测结果如下图所示,DTX和THP的出峰时间分别为5.514 min和7.218 min,峰形良好,且载体材料对DTX和THP的色谱峰均无影响。两药专属性分别见图1~2。

A.DTX标准品;B.MSNs;C.THP-DTX-MSNs
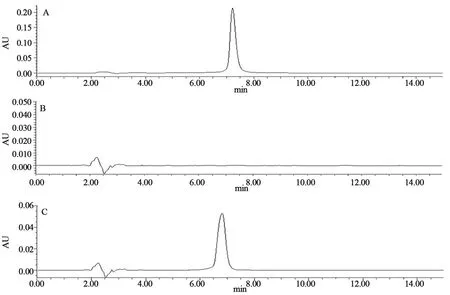
A.THP标准品;B.FA-PDA-MSNs;C.FA-PDA-THP-DTX-MSNs
2.2.3 标准曲线的绘制精密配制浓度为2.00、10.00、20.00、50.00、80.00、100.00 μg·mL-1的DTX标准溶液。按“2.2.1”项下的色谱条件进行测定,以峰面积(As)为纵坐标,以DTX浓度(C)横坐标进行回归分析,得标准曲线方程为:As=10 679.333 5C-18 389.564 1(R2=0.999 0),结果表明,DTX在2.00~100.00 μg·mL-1范围内线性关系良好。
精密配制浓度为1.78、4.44、8.88、22.20、35.52、44.40 μg·mL-1的THP标准溶液。按“2.2.1”项下的色谱条件进行测定,以峰面积(As)为纵坐标,以浓度(C)为横坐标进行回归分析,得标准曲线方程为:As=48 217.862 7C-69 630.331 8(R2=0.999 0)。结果表明,THP在1.78~44.40 μg·mL-1范围内线性关系良好。
2.2.4 精密度考察分别取低、中、高(10.00、50.00、100.00 μg·mL-1)浓度的DTX供试品溶液与低、中、高(4.44、22.20、44.40 μg·mL-1)浓度的THP供试品溶液,按“2.2.1”项下条件,每种样品分别进样6次,连续测定3 d,测定并记录峰面积,计算DTX与THP日内精密度及日间精密度,结RSD均小于2.00%,此方法的日内、日间精密度良好,符合含量测定的要求。
2.2.5 加样回收率的测定取1.0 mg·mL-1MSNs乙醇溶液1 mL分别与低、中、高(10.00、50.00、100.00 μg·mL-1)浓度的DTX供试品溶液或低、中、高(4.44、22.20、44.40 μg·mL-1)浓度的THP供试品溶液以1∶1体积比进行混合。按照“2.2.1”项下的色谱条件进行测定计算其药物含量。回收率计算公式为:回收率=(测定量-样品量)/加入量×100%。结果可知,两药加样回收率均满足《中国药典》2020年版对于含量测定的要求。
2.3 药物的负载以及包封率与载药量检测方法的建立称取定量MSNs置于10 mL的载药溶剂中,超声至分散均匀,加入定量THP和DTX溶解,避光室温搅拌数小时实现两药同时负载。
利用超声溶解法对FA-PDA-THP-DTX-MSNs载药量和包封率进行测定。即精密称量少量FA-PDA-THP-DTX-MSNs放置于无水乙醇(1 mL)中,超声30 min,8 000 r·min-1离心5 min,得上清,沉淀继续加入无水乙醇(1 mL)超声,重复3次。合并上清,以“2.2.1”项下液相条件对两种药物进行含量测定,计算载药量、包封率,结果见表1。

表1 FA-PDA-THP-DTX-MSNs载药量包封率测定结果(n=3)
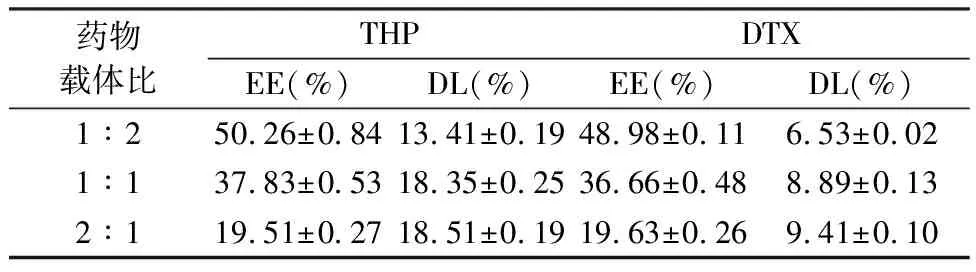
表2 药物载体比对两药包封率及载药量的影响(n=3)
DL(%)=W载/W总×100%
EE(%)=W载/W投×100%
其中DL为载药量,EE为包封率,W总为载药载体的质量,W载为载药载体中药物的质量,W投为投药量。
结果显示,利用超声溶解法测定FA-PDA-THP-DTX-MSNs中DTX和THP的包封率、载药量准确度较高,重现性较好,可利用此法对纳米给药系统的包封率和载药量进行测定。
2.4 制备工艺优化
2.4.1 药物载体比例的选择为了优化两药的负载条件,本部分对药物载体比以及载药时间分别进行了单因素考察。固定载药时间为12 h,改变药物载体比,两药包封率和载药量结果显示随着MSNs载体的比例减小,两种药物的包封率均减小。当药物载体比在1∶1~2∶1之间时,载药量仅有少量增加。造成这一现象的原因可能是由于当药物载体比达到1∶1时,MSNs孔道内药物几乎达到饱和。因此选择药物载体比为1∶1。
2.4.2 载药时间的选择固定负载溶剂为25%乙醇溶液,药物载体比为1∶1,改变载药时间,两药包封率和载药量结果如表3。结果显示载药时间在6~12 h时,随着搅拌时间的增加,两种药物的载药量和包封率均显著增加,当载药时间在12~18 h时,两种药物的总载药量仅有少量增加,这与在考察药物载体比中所提到的MSNs孔道所能负载药物达到饱和原因一致。两种药物随着搅拌时间的增加,两种药物的进入MSNs孔道的比例时有所变化,这一现象产生的原因是由于两种药的结构性质不同对于MSNs的吸附能力有所不同,因此存在一定的竞争关系。为了保证两种药物的最佳比例,选择搅拌时间为12 h作为载药时间。

表3 载药时间对两药包封率及载药量的影响(n=3)
2.4.3 聚多巴胺膜的包覆及条件优化聚多巴胺膜包覆基本操作如下:称取定量10.00 mg THP-DTX-MSNs置于10 mL 10 mmol·L-1的Tris-HCl溶液中(pH=8.5)超声分散均匀,后向其中加入定量盐酸多巴胺,室温搅拌数小时,8 000 r·min-115 min离心得聚多巴胺膜包覆的载药介孔二氧化硅纳米粒(PDA-THP-DTX-MSNs)[16]。
首先固定包覆时间为3 h,向10 mmol·L-1的Tris-HCl(pH=8.5)溶液加入盐酸多巴胺,使盐酸多巴胺终浓度分别为0.25、0.50、0.75、1.00、1.25 mg·mL-1。反应结束后离心得PDA-THP-DTX-MSNs并使用激光粒度仪测定粒径。
图3结果表明,在盐酸多巴胺浓度为1.0 mg·mL-1时系统PDI增加到0.657左右,表明测定溶液体系中粒径大小分布十分不均匀,造成这种现象可能是由于溶液中盐酸多巴胺浓度过大,部分盐酸多巴胺自身氧化成球状纳米粒,而这种纳米粒与PDA-THP-DTX-MSNs的粒径相差较为悬殊,最终使得体系内的PDI分散系数增加。另外为尽可能实现PDA对于MSNs孔道内药物的封堵作用,选择盐酸多巴胺浓度为0.50 mg·mL-1作为包覆浓度,此时纳米粒子的粒径约为183.5 nm,既可实现对于孔道内药物的封堵又满足纳米粒子被动靶向的粒径要求。

图3 盐酸多巴胺浓度对于聚多巴胺膜包覆MSNs粒径的影响(n=3)
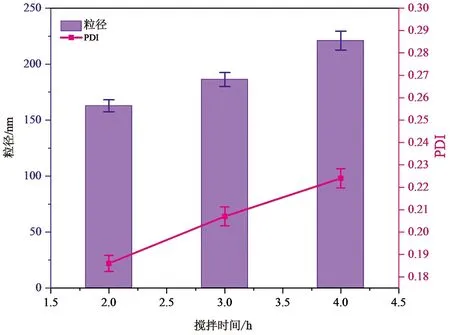
图4 搅拌时间对于聚多巴胺膜包覆介孔二氧化硅粒径的影响(n=3)
在确定盐酸多巴胺浓度的基础上,分别搅拌2、3、4 h。反应结束后离心得PDA-THP-DTX-MSNs。取少量利用去离子水超声分散均匀,激光粒度仪测定粒径。
结果显示,随着搅拌时间的增加,纳米粒子的粒径逐渐增加,PDI分散系数在固定盐酸多巴胺浓度的条件下,无明显变化。为同时满足封堵及被动靶向作用,故选择搅拌时间为3 h,此时粒径约为186.5 nm。
2.5 叶酸的偶联称取1.0 mg PDA-THP-DTX-MSNs超声分散于200 μL浓度为20 mg·mL-1的聚乙烯亚胺(PEI)溶液中,搅拌2 h,得到PEI功能化的纳米颗粒。随后将4 mL浓度为1 mg·mL-1的1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)和10 mL浓度为1 mg·mL-1的N-羟基丁二酰亚胺(NHS)溶液加入1 mL FA溶液(1 mg·mL-1二甲基亚砜中),反应30 min,以活化FA的羧基。将该溶液快速加入PEI功能化的PDA-THP-DTX-MSNs溶液中搅拌12 h,最后在8 000 r·min-1离心10 min,水和乙醇洗涤,收集FA偶联的PDA-THP-DTX-MSNs纳米粒子(FA-PDA-THP-DTX-MSNs),30 ℃条件下,鼓风干燥,最终得FA-PDA-THP-DTX-MSNs干燥粉末。
2.6 FA-PDA-THP-DTX-MSNs纳米粒的表征
2.6.1 纳米粒的粒径与电位由PDA-THP-DTX-MSNs透射电镜图(见图5)可知,PDA-THP-DTX-MSNs的粒径在MSNs的基础上有所增加,在MSNs的外层有明显的黑色薄膜形成。图6显示MSNs、PDA-THP-DTX-MSNs与FA-PDA-THP-DTX-MSNs 3种纳米粒粒径依次增加,PDI指数均小于0.3,具有良好的分散性。

A.MSNs;B.PDA-THP-DTX-MSNs
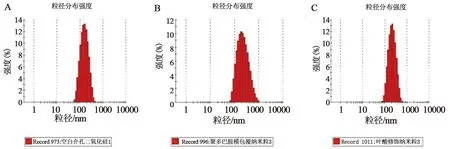
A.MSNs;B.PDA-THP-DTX-MSNs;C.FA-PDA-THP-DTX-MSNs
图7显示PDA-THP-DTX-MSNs因PDA表面羟基的存在[17]其Zeta电位在MSNs的基础上进一步降低为(-24.77±1.15)mV,由于PEI胺基的质子化,使得PEI-PDA-THP-DTX-MSNs电位反转(+38.03±0.54)mV,最后活化后FA的羧基与PEI的胺基通过酰胺反应进行连接。由于FA共轭作用以及对PEI胺基的质子化覆盖作用,使得FA-PDA-THP-DTX-MSNs的Zeta电位略微下降至(+23.3±0.67)mV[18]。
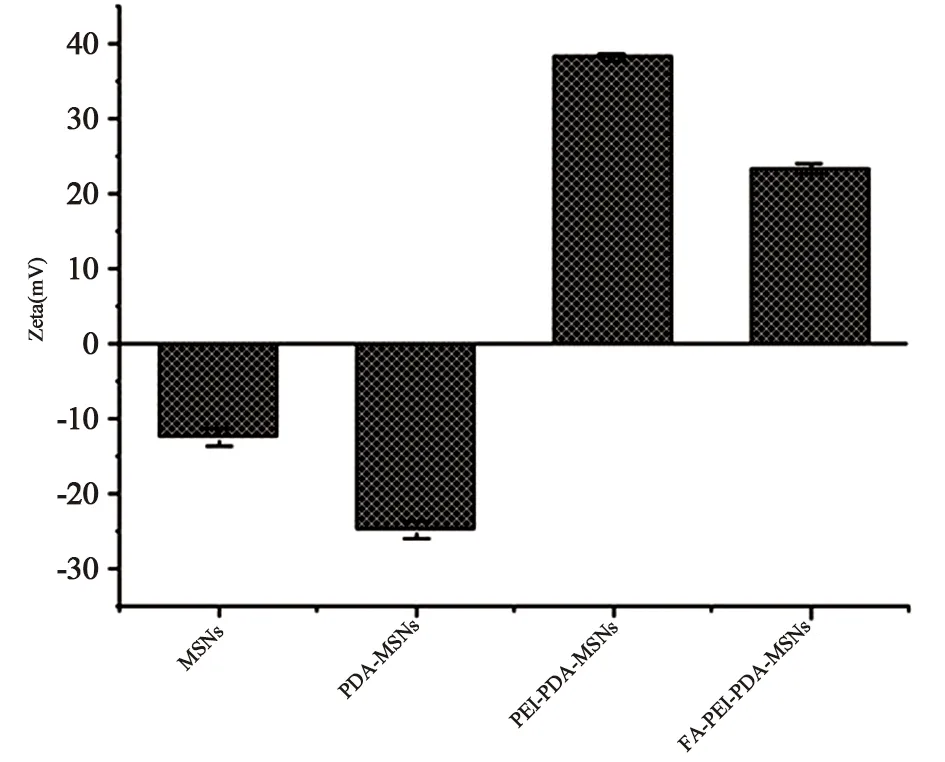
图7 MSNs、PDA-THP-DTX-MSNs、PEI功能化PDA-THP-DTX-MSNs、FA-PDA-THP-DTX-MSNs Zeta电位(n=3)
2.6.2 N2吸附-解吸附分析N2吸附-解吸附分析试验结果如图8~9所示,PDA-MSNs及MSNs两者的物理吸附-脱附等温线均为典型的IV型脱附曲线。通过BET法和BJH法对PDA-MSNs的平均比表面积、孔容和孔径进行计算分析,(比表面积为325.795 5 m2·g-1;孔容积为0.520 9 cm3·g-1;孔径为3.11 nm)结果表明孔径大小没有变化,但是比表面积和孔容积均明显减小,由此可得PDA可实现对于MSNs的封堵作用。

图8 MSNs、PDA-MSNs氮气吸附-解吸附等温图
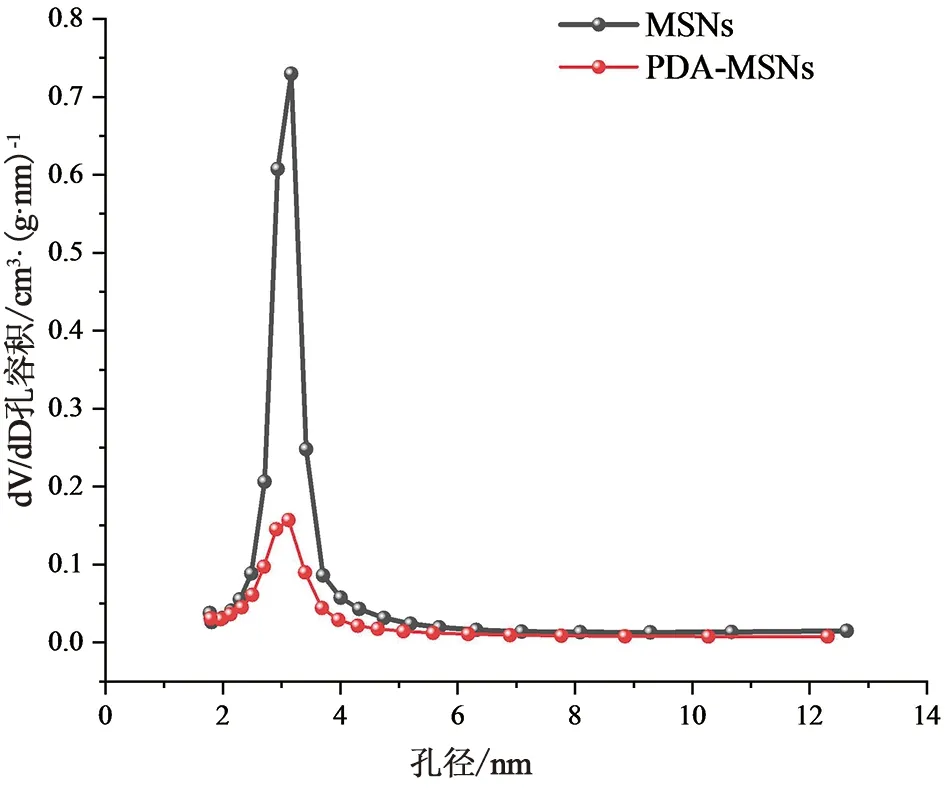
图9 MSNs、PDA-MSNs孔径分布图
2.6.3 傅立叶红外变换光谱(FTIR)MSNs、PDA-MSNs、FA-PDA-MSNs以及FA的傅立叶红外光扫描图谱如图10所示,1 087 cm-1处为Si-O-Si反对称伸缩振动吸收峰,在966 cm-1处为Si-OH的弯曲振动吸收特征峰。808 cm-1为Si-O键的对称伸缩振动吸收峰,其上均为MSNs的特征红外峰。除FA单体外,其他样品的红外特征图谱均在MSNs特征图谱上增加其特征峰,3 814、1 541、1 238 cm-1等作为PDA苯环上的特征峰,在MSNs原有吸收的基础上有一定程度的叠加,而在未共有波长处则由于PDA对MSNs进行了覆盖,MSNs特征吸收有所减弱。1 622 cm-1和1 608 cm-1分别作为酰胺键的特征峰以及FA的红外特征图谱,均可作为FA成功偶联的判断依据[19-20]。
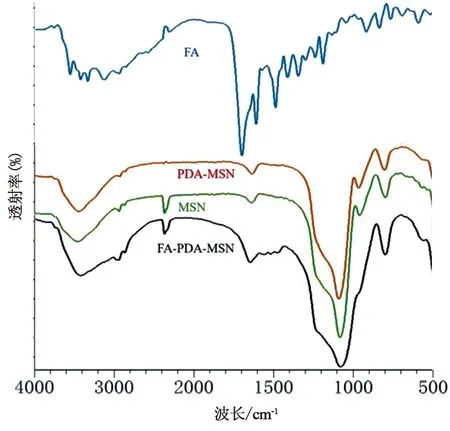
图10 MSNs、PDA-MSNs、FA-PDA-MSNs以及叶酸的傅立叶红外扫描图谱
2.6.4 热重分析(TG)图11为热重分析图,图中由上至下分别表示MSNs、PDA-MSNs和FA-PDA-MSNs的热重曲线,结果显示当检测温度从室温升至800 ℃时,三者的失重百分率分别为8.5921%、21.0634%以及28.8021%。MSNs在200 ℃以下重量的损失主要是由于高温脱去其表面吸附的水;200 ℃至800 ℃之间主要是由于是制备过程中模板剂CTAB未被彻底煅烧所造成的[21]。扣除模板剂和吸附水的失重后,PDA-MSNs、FA-PDA-MSNs在200 ℃至800 ℃之间的失重率分别为12.4713%和20.2100%。因此,该纳米给药系统中按照优化后的条件PDA和FA靶向基团的接枝率分别为12.4713%和7.4687%。
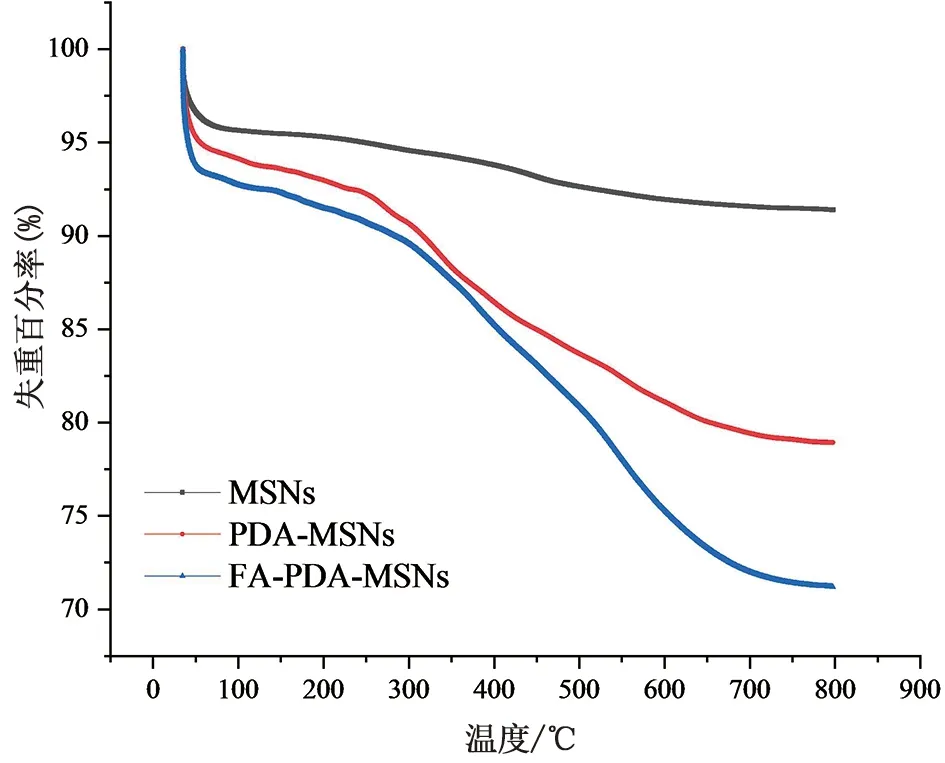
图11 MSNs、PDA-MSNs、FA-PDA-MSNs热重分析曲线
2.7 体外释药方法与结果
2.7.1 体外释药试验结果将透析袋中装入2 mL FA-PDA-THP-DTX-MSNs 1% Tween-80 PBS以及相同剂量的THP-DTX游离溶液(各透析袋中THP、DTX含量尽量保持一致)分别放置于条件为pH 5.0、pH 7.4、pH 5.0+NIR、pH 7.4+NIR的环境中(n=3)。在释药开始后,分别于0.25、0.5、1、2、3、4、6、8、10、12、24、48、72、96 h量取1 mL释药介质同时补加等量同温空白介质(激光照射组照射条件为2 W·cm-2,照射5 min后,后续同以上操作),计算药物的累积释放率Q(%)。不同条件下DTX及THP累计释放百分率以及体外释放曲线见图12~13。各时间点药物累积释放百分率(Q)的计算公式为:

图12 DTX和纳米粒在各条件下的体外释放曲线(n=3)

图13 THP和纳米粒在各条件下的体外释放曲线(n=3)
式中Ct为在各时间点测得释放介质中的药物浓度(mg·mL-1),W为投入药物的总重量(mg),V0为释放介质的总体积,V为每次取样的体积。
两药的累计释放曲线如图12~13所示,游离组在0~6 h快速释放,累计释放率可达到85%左右,且不随pH值的变化而发生变化。而FA-PDA-THP-DTX-MSNs中的DTX及THP的释放相较于游离药物能够显著延长体外释放时间,并且均表现出明显的pH响应。药物在96 h的累计释放结果显示,FA-PDA-THP-DTX-MSNs在pH=5.0的酸性环境下释放量可达至63.72%±1.66%,pH=7.4的中性环境下仅为16.57%±0.51%。这是由于在酸性条件下PDA会部分分解,有利于药物的释放。除此之外,分别在6、12、24、48、72 h时间点利用2 W·cm-2的NIR辐照5 min,结果显示两种药物在NIR照射的条件下其释放量均有一定程度的提高,且在酸性条件下,提高幅度更大约达20%。由此可见在酸性环境和近红外激光的条件下可加快靶向纳米给药系统内药物的释放,减少化疗药物非特异性释放。
2.7.2 方程拟合依据上述两药在不同条件下各时间点下的累计释放百分率,对FA-PDA-THP-DTX-MSNs的体外释放曲线进行方程拟合,以进一步阐明FA-PDA-THP-DTX-MSNs的释药机制。
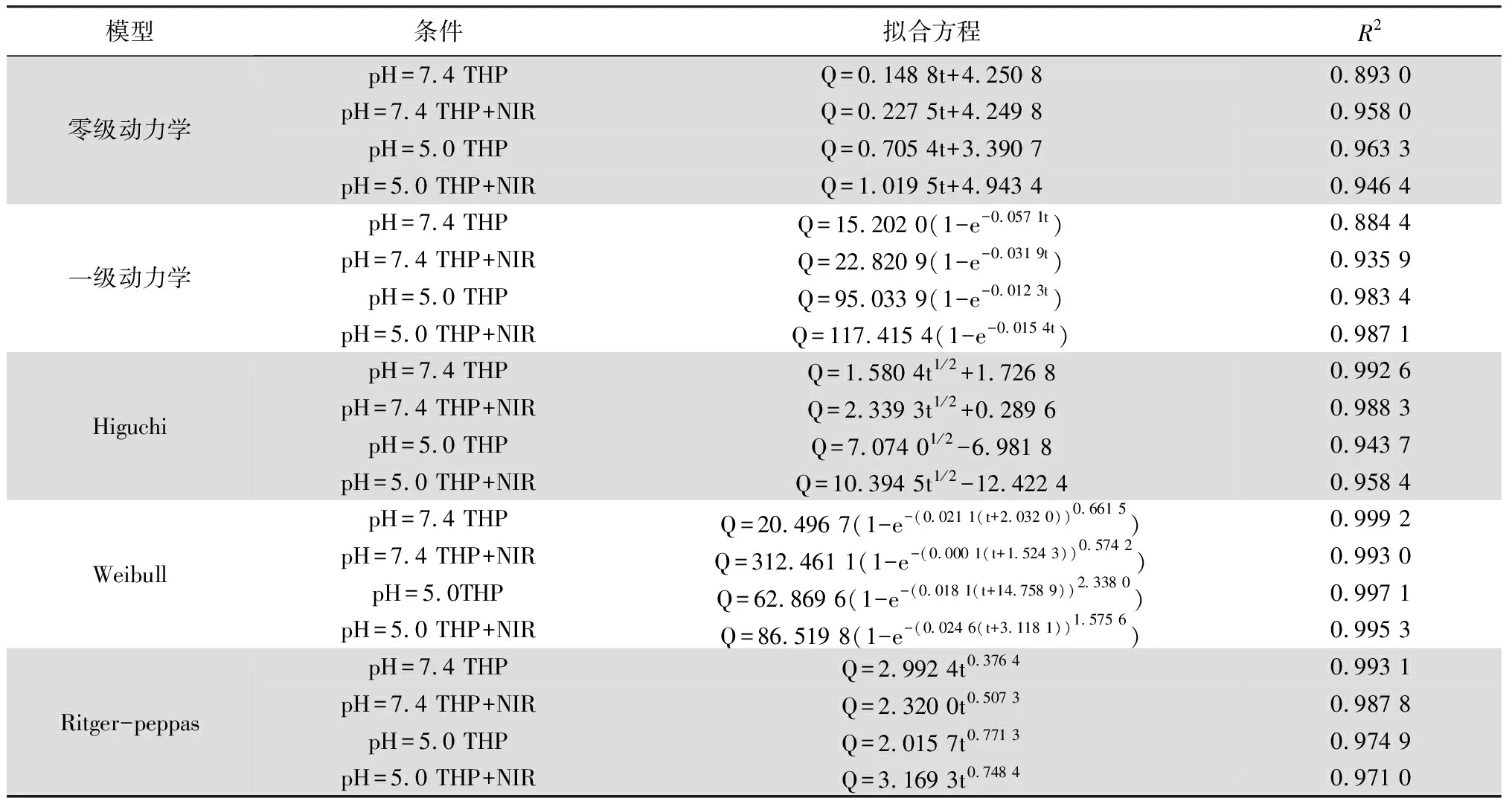
体外释放曲线方程拟合结果显示(见表4~5),两种药物无论是在pH为7.4还是5.0,以及是否有NIR照射的条件下,其释药曲线均与Weibull释药模型的拟合度最高,该模型可较好的适用于速释和缓释药物的释放拟合。为进一步研究其释放机理,将不同条件下的药物释放曲线与Ritger-Peppas方程Q=Ktm(K、m为常数)拟合。当m≤0.45时,其药物释放机制为Fick扩散;当0.45 表4 DTX累积释药曲线的拟合方程 表5 THP累积释药曲线的拟合方程 本试验中以MSNs作为纳米药物载体,DTX和THP作为模型药物,负载于MSNs孔道及表面,利用盐酸多巴胺在碱性条件下氧化形成PDA薄膜包覆于MSNs表面,最后利用酰胺反应将FA靶头修饰于PDA薄膜的表面成功制备得FA-PDA-THP-DTX-MSNs纳米粒。对FA-PDA-THP-DTX-MSNs纳米粒的制备工艺进行优化,考察了优化后纳米粒的表征以及释药行为,证实了靶向纳米粒的成功制备并能实现刺激性响应释药。综上所述,本研究从药剂学的角度为实现化疗与光疗相结合的靶向治疗方法提供了新的策略和依据。基于光热效应的聚多巴胺膜靶向纳米粒作为一种新型的可实现光热治疗与化疗相结合的纳米粒,展现出良好的应用前景,可为日后实现光热治疗和化疗联用提供一定的参考。但与此同时,新的递送系统在临床应用方面还存在着各方面的困难,具体例如其表面正电荷极易与血液中的组分发生聚集而导致生物毒性增强和生物利用度降低,可使用如透明质酸或中药多糖进行逆转电荷。因此在后续实验中,FA-PDA-THP-DTX-MSNs纳米递送系统可进行生物安全性以及体内外药效学实验,对其进行进一步优化。
3 结论
猜你喜欢
疯狂英语·新悦读(2023年2期)2023-10-12
读者(2022年9期)2022-04-22
环境卫生工程(2021年4期)2021-10-13
婚姻与家庭·婚姻情感版(2021年6期)2021-06-01
疯狂英语·新读写(2020年3期)2020-06-06
时代英语·高一(2019年5期)2019-09-03
天然产物研究与开发(2016年6期)2016-06-05
海峡科技与产业(2016年3期)2016-05-17
小说月刊(2015年6期)2015-12-16
化工进展(2015年3期)2015-11-11
