
蕨类植物叶绿体基因ycf94的分子进化研究
2023-08-05 03:51李子菲苏应娟王艇
热带亚热带植物学报 2023年4期
李子菲, 苏应娟, 王艇*
蕨类植物叶绿体基因的分子进化研究
李子菲1, 苏应娟2,3*, 王艇1*
(1. 华南农业大学生命科学学院, 广州 510642; 2. 中山大学生命科学学院, 广州 510275; 3. 中山大学深圳研究院, 深圳 518057)
基因是近年来在叶绿体基因组中新发现的一个基因,在蕨类植物中表现高度保守。该研究共选取94种蕨类植物,在系统发育背景下,对基因的结构特征、密码子偏好性、进化速率和适应性进化进行分析。结果表明,基因的密码子偏好性较弱,偏好使用以 A/U 结尾的密码子,且不同物种间的偏好性存在一定差异。密码子偏好性的形成主要受到突变压的影响,同时也存在其他因素的作用;基于凤尾蕨科和其他蕨类中基因的结构特征存在区别,对两者的分子替换速率进行了比较,表明颠换率、非同义替换率和值间存在显著差异;仅检测出1个正选择位点74A,强烈的负选择作用表明基因的结构和功能基本趋于稳定。这为蕨类系统发育分析提供了新依据,并提供了解析基因功能的线索。
蕨类植物;;密码子偏好性;进化速率;适应性进化
叶绿体基因组具有长度短、拷贝数高、保守性强的特点,长期被广泛用于植物系统发育和分子进化研究[1]。叶绿体基因组作为双链环状闭合DNA,大小一般为110~116 kb,具有典型的四区结构,包含了大单拷贝区(large single copy, LSC)、小单拷贝区(small single copy, SSC)和2个反向重复区(inverted repeats, IR)。目前普遍认为陆地植物叶绿体基因组的基因组成是大致相似的,大约有80个蛋白编码基因(CDS, protein-coding gene)、30个tRNA基因和4个rRNA基因[2]。其中也包含一些保守的开放阅读框(open reading frame, ORF),其所编码的蛋白相关功能仍在摸索中,通常称为假定叶绿体开放阅读框(hypothetical chloroplast open reading frame)基因()。
蕨类作为最早脱离水体适应陆生环境、进化出维管组织的植物类群,在整个陆地植物的起源和演化历史研究中有着重要的地位[3]。蕨类植物现存约有12 000种,它在形态上具有丰富的多样性,广泛分布在热带和亚热带地区。同时,蕨类的质体基因组有独特的结构,相比于与种子植物的质体基因组结构基本一致的蕨类祖先类型“莲座蕨型”,“铁线蕨型”分别在跨IR/LSC区和LSC区中发生了约20和3 kb的两次重叠倒位,而“铁线蕨型”占据了蕨类90%的种类[4]。2016年发表的PPG I (the pteridophyte phylogeny group Ⅰ)系统是一个基本建立在分子系统发育研究之上的现代蕨类植物分类系统[5],其中描述的分类较为全面且被接受。目前,有关蕨类叶绿体基因组和分类系统等各类信息仍在不断完善中。
Song等[6]于2018年在薄囊蕨类中发现1个新的假定ORF,并将其命名为。研究表明,位于K和16之间,在薄囊蕨类中表现高度保守。同时在蕨类、石松类和苔藓类的质体中都发现其同源性,而种子植物中不存在该序列。在陆地植物中质体的进化历史是动态的,其基因组存在基因丢失的现象,基因会从质体基因组转移到核基因组[7]。或许存在于陆生植物的祖先质体中,随后在种子植物中发生基因丢失。
根据序列的保守程度,以及其编码跨膜蛋白和具有多个RNA编辑位点的特征[6],可以表明是一个具有功能意义的蛋白编码基因[8],但其起源和功能目前尚不明确。本研究以94种蕨类植物的基因为研究对象,通过分析的碱基组成和密码子偏好性,并在系统发育背景下,分析其进化速率和选择压力,来探究的基因结构特征和在蕨类植物中的进化样式,挖掘在叶绿体基因组系统发育关系分析中的可用性,为蕨类植物分类提供新的依据,为进一步研究的起源和功能等提供基础信息。
1 材料和方法
1.1 提取序列数据
根据的基因序列作为参考序列,用NCBI-BLAST在线比对(E value<1e-6)在GenBank数据库上选取了94种蕨类植物叶绿体全基因组,涵盖了3目19科60属(附录1)。采用Genious prime软件[9]提取基因,并使用MEGA X软件通过MUSCLE (密码子)模块进行序列比对并对结果进行手工校正。
1.2 构建系统发育树
进行94种蕨类植物基于K+L串联数据集的多基因系统发育分析,构建3种系统发育树。PhyloSuite v1.2.2软件[10]导入94条序列并提取K和L基因,在MATTF插件上序列比对后进行基因串联。用ModelFinder插件来选择最佳的核苷酸替代模型GTR+F+I+G4,以6种合囊蕨目作为外类群, 利用MrBayes方法重建贝叶斯(bayesian inference, BI)树。并且用MEGA分别构建邻接(neighbor-joining, NJ)树和最大似然(maximum likehood, ML)树。综合分析3种树,结合PPGI系统发育关系进行手工调整,得出系统发育树。
1.3 密码子偏好性分析
以94种蕨类植物的基因为对象,使用在线软件CUSP计算密码子第一、第二、第三位的GC含量(分别用GC1、GC2、GC3表示)和总GC含量;CodonW v1.4.2软件统计同义密码子相对使用度(RSCU, relative synonymous codon usage)和有效密码子(ENC, effective number of codon)。使用软件SPSS 24对数据进行Pearson相关性分析。
GC3-GC12分析 密码子的使用受到碱基的突变压和自然选择的双重作用[11],通过分析GC12和GC3的相关性,研究密码子3个位置上的碱基组成是否相似和影响密码子偏好性的主要因素。当GC12和GC3之间显著相关,说明密码子3个位置上的碱基组成无差异,密码子的使用主要受突变的影响;当GC12和GC3之间相关性不显著, 说明密码子第一、第二和第三位碱基组成不相似, 密码子的使用主要受自然选择的影响。
ENC-plot分析 分析GC3和ENC的相关性,研究碱基组成对密码子偏好性的影响。以GC3为横坐标、ENC值为纵坐标绘制散点图,并绘制ENC期望值标准曲线(ENC=2+GC3+29/[GC32+(1–GC3)2])进行比较。当基因散点分布在标准曲线附近,说明密码子偏好性受突变影响较大;而散点位于曲线较远下方位置时,密码子偏好性受自然选择影响较大。
最优密码子 ENC值反映了同义密码子非均衡使用的偏好程度,取值范围为20~61。通常基因表达量越高,密码子偏好性越强,ENC值越小[12]。分析94种蕨类植物基因主要偏好密码子, 构建以ENC值为标准的高低表达库(选择ENC值最低和最高的10个基因),计算RSCU值(ΔRSCU= RSCUHigh–RSCULow),选择RSCU>0.08且RSCU>1的作为最优密码子。
1.4 分子进化速率分析
采用HyPhy v2.2.4软件[13]基于核苷酸模型HKY85和密码子模型MG94×HKY85_3×4计算各物种分支基因的转换率(transition rate, trst)、颠换率(transversion rate, trsv)、转换率和颠换率的比值(trst/trsv)、非同义替换率(nonsynonymous substi- tution rate, dN)、同义替换率(synonymous substitu- tion rate, dS),以及非同义替换速率和同义替换速率的比值(=dN/dS)。通过软件SPSS 24使用Mann- Whitney秩和检验对数据进行相关性分析。
1.5 适应性进化分析
使用PAML v4.9软件[14]中的codeml程序检测基因中是否存在氨基酸正选择位点。采用检测谱系之间选择压力的3种模型:分支模型(Branch model)、位点模型(Site model)和分支-位点模型(Branch-site model)。
分支模型中,单比率模型(one-ratio, M0)假定所有分支的值相同;自由比率模型(free-ratio, F)假定所有分支的值均不相同;二比率模型(two-ratio, M2)假定前景支与背景支的值不相同。通过似然比检验进行模型比较,M0和检验不同分支间值的差异性,M0和M2检验前景支和背景支间值的差异性。
位点模型假定不同位点的值不同,其中存在4对比较模型:M0 (one-ratio)和M3 (discrete)检测位点间值是否一致;M1a (neutral)和M2a (selection), M7 (beta)和M8 (beta&),M8和M8a (beta&=1)用于检测是否存在正选择位点。
分支-位点模型假定了分支间和位点间值都存在差异。对模型Model A和零假设进行比较,检测前景支中的部分位点是否受到正选择作用。
2 结果和分析
2.1 ycf94的基因特征分析
取样的94种蕨类植物基因长度为222~156 bp。其中可以大致分为2种类型:以起始密码子为T(ACG)开头发生C to U RNA编辑的序列共28条,其中包括全部的凤尾蕨科27条,大多为219 bp (共20条);起始密码子为M(AUG)的序列共66条, 包含除凤尾蕨科外的18科,大多为216 bp (共50条),其中6条合囊蕨目的大小均为177 bp。
2.2 94种蕨类植物的系统发育树
利用K+L串联基因构建NJ、ML和BI3种系统发育树并对其进行比较,用蕨类分类系统PPGI进行修正。
在ML和BI中水龙骨亚目和铁角蕨亚目为姐妹群且都是单系,与PPGI系统一致;而NJ中水龙骨亚目属于多系。BI支持铁角蕨科和肠蕨科为姐妹群, ML支持铁角蕨科位于铁角蕨亚目的基部位置。BI与PPGI系统最为接近,所以最后以BI为标准。构建出的系统发育树(附录2)主要为后续研究分析提供系统发育背景。
2.3 密码子偏好性分析
2.3.1 密码子碱基组成分析
附录3为94种蕨类基因的密码子碱基组成,其中GC含量为23.73%~47.75%,GC1、GC2和GC3分别为19.72%~47.22%、23.94%~47.22%和20.34%~56.94%,说明密码子总体的碱基组成偏向A/U。ENC值为61~34.89,在近缘物种中基因的密码子偏好性存在差异。其中有19条序列的ENC=61即每个密码子都被均衡使用。ENC值以35作为判断密码子偏好性强弱的标准[15],大部分的密码子偏好性较弱。
对基因的密码子相关参数进行热图统计,大多数的GC3要高于GC1、GC2和GC值;合囊蕨目的GC各项数值要低于其他蕨类,凤尾蕨科的GC1和GC2总体上也低于其他蕨类(图1)。
由表1可见,GC1、GC2、GC3和GC相互间均为极显著相关,表明基因密码子三位的碱基组成相似;而ENC值与各项GC值相关性不显著,碱基组成对密码子偏好性的影响较小。

表1 ycf94密码子各参数的相关性分析
*:<0.05; **:<0.01

图1 ycf94基因的密码子相关参数热图统计
2.3.2 GC3-GC12分析
由图2可见,大部分散点位于对角线之下,除天星蕨()外,大部分物种的基因的GC3值大于GC12。相关性分析结果表明,双尾检验=0.00 (<0.01),相关系数为0.739, GC3和GC12相关性极显著,说明密码子偏好性主要受到突变的影响。
2.3.3 ENC-plot分析
由图3可见,散点的分布较为分散,且一部分落在距离曲线较远的位置,则该部分基因密码子偏好性受选择影响较大。有40条序列的ENC比值为–0.1~0.1,表明ENC值与期望值相差较小,该部分基因的密码子偏好性受突变影响较大。
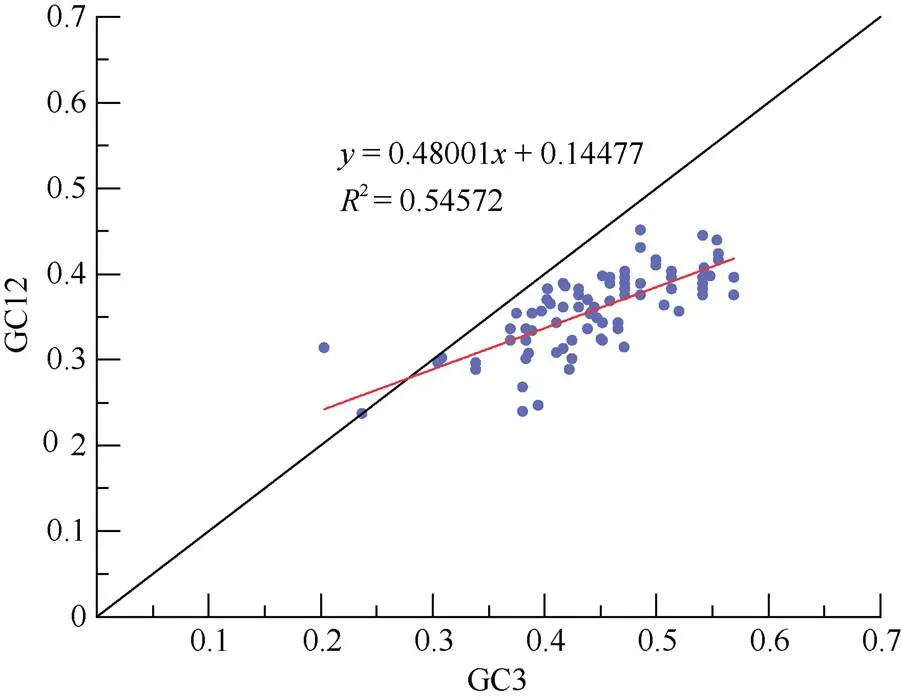
图2 中性绘图
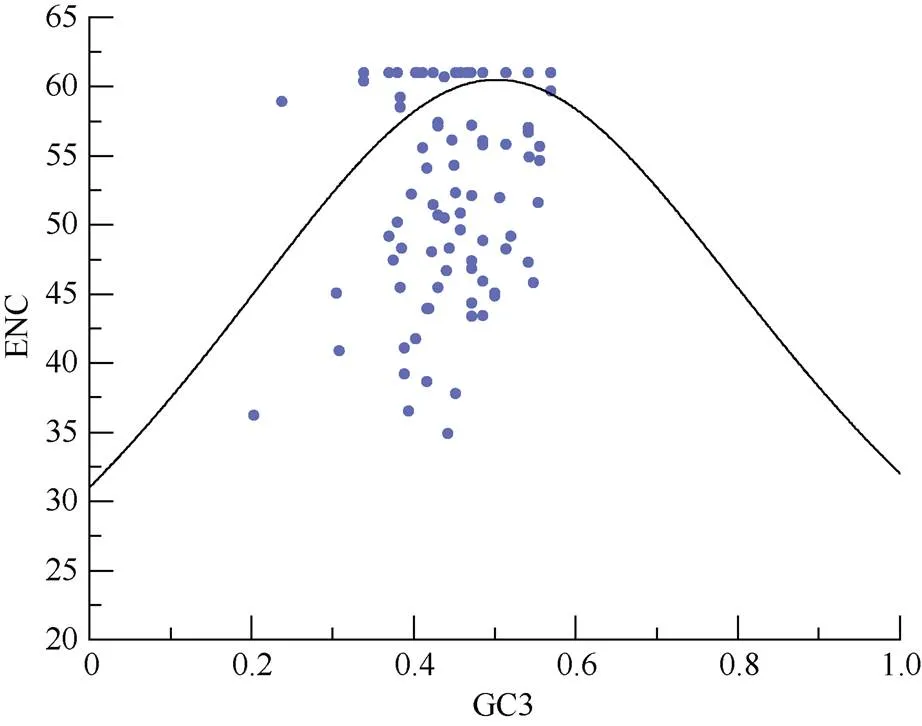
图3 ENC-plot绘图
2.3.4最优密码子
从表2可见,共筛选出高频密码子(RSCU>1) 26个,高表达密码子(△RSCU>0.08) 24个,而同时满足这两个条件的最优密码子有14个:UUU、GUU、GUA、GUG、UCU、CCC、ACC、GCC、UAU、AAA、GAU、AGG、GGC、GGA。其中8个以A/U结尾,6个以G/C结尾。
2.4 进化速率分析
对凤尾蕨科和其余蕨类的替换速率进行差异显著性分析,数据表明(附录4),凤尾蕨科除了同义替换率外的进化速率均值都大于其他蕨类(图4),秩和检验中颠换率,非同义替换率和的值分别是0.011、0.017和0.036 (<0.05),说明凤尾蕨科和其他蕨类基因的trsv、dN和存在显著差异, 而trst、trsv/trst和dS无显著差异。

表2 ycf94基因的密码子RSCU统计以及最优密码子
*: 最优密码子; **: 高表达密码子; ***: 高频密码子。
*: Optimal codon; **: High expression codon; ***: High frequency codon.
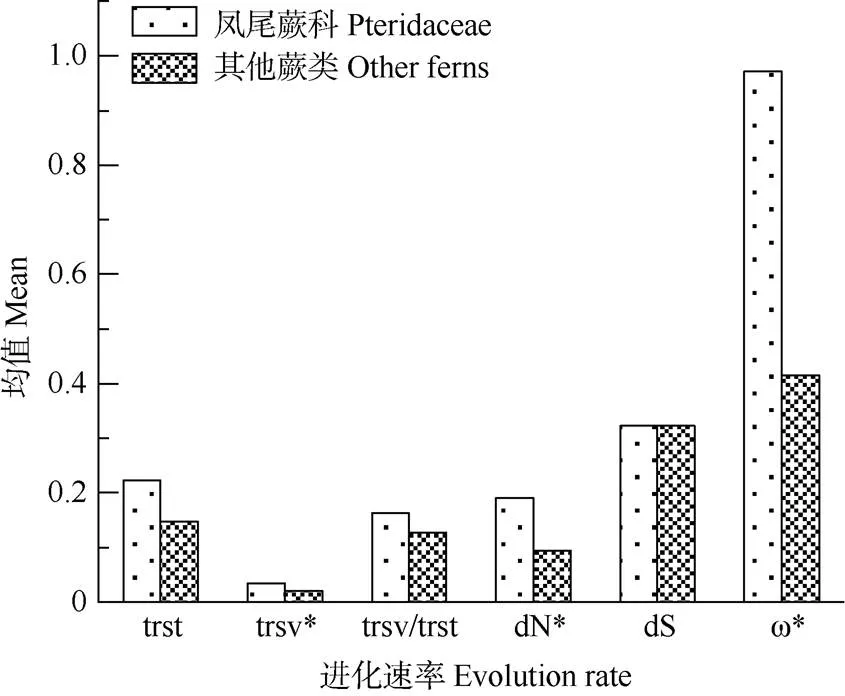
图4 凤尾蕨科与其他蕨类ycf94进化速率均值比较及Mann-Whitney检验
2.5 适应性进化分析
表3和4是PAML得出的基因适应性进化分析的结果。对各模型下计算出的对数似然值进行模型间的似然比检验,分析显著性选择更为适合的模型。结果表明,分支模型下M0和检验结果显著(=0.024),表明不同分支间值存在显著差异。二比率模型中设定了以凤尾蕨科为前景支,其他蕨类为背景支,M0和M2检验结果极显著(=6.188× 10–6),表明凤尾蕨科和其他蕨类的值存在极显著差异。
位点模型下,结果显示位点间值存在显著差异(=0.000),且3对模型比较都显示基因中存在正选择位点。以BEB后验概率大于95%为标准,模型M2a和M8均鉴定出1个正选择位点74A。
在分支-位点模型中,同样以凤尾蕨科为前景支,似然比检测结果显示零假设模型和Model A间不存在差异(=1.000),故不接受模型A的结果。
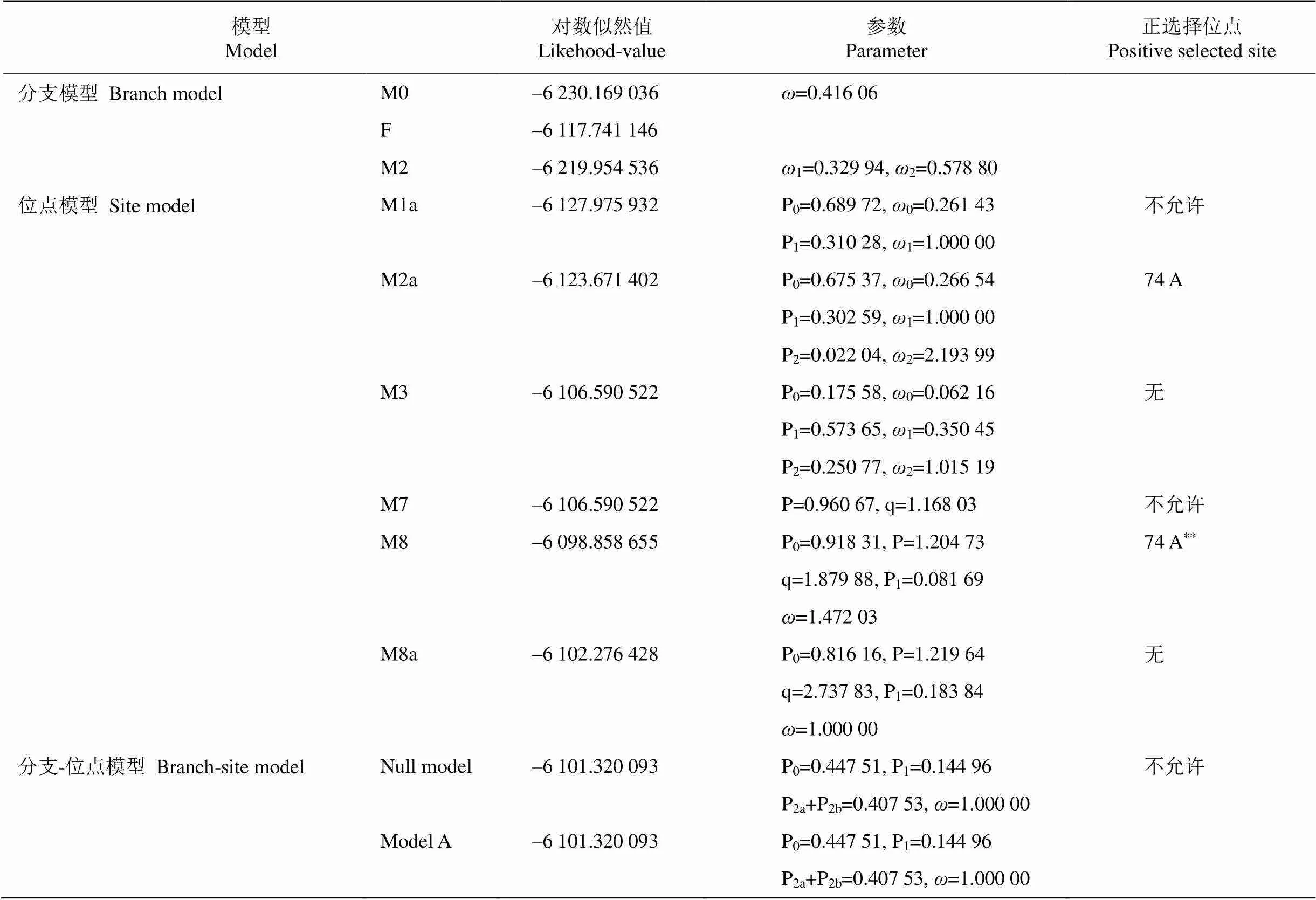
表3 不同模型下的参数估计值和对数似然值
**: 后验概率<99%
**: Posterior probability <99%

表4 似然比检验
*:<0.05; **:<0.01
3 结论和讨论
基因在蕨类中表现出高度保守性,但在种子植物中没有发现同源序列这个重要特征,可能成为系统发育分析中的一个新的分子标记。同时,研究发现在蕨类中存在2种结构样式:起始密码子ACG发生RNA编辑的、基因长度多为219 bp的序列和以起始密码子为AUG、基因长度多为216 bp的序列。其中全部的凤尾蕨科基因序列都表现为前一种模式,以及三叉蕨科的也属于同一结构,其余的序列均为后者。因此,的起始密码子发生C to U RNA编辑虽然不属于凤尾蕨科的独有结构,但RNA编辑作为表观遗传现象之一拥有可遗传性,推测凤尾蕨科的共同祖先中基因起始密码子存在RNA编辑,该基因结构是凤尾蕨科的共衍征。
编译氨基酸过程中同义密码子的使用是存在偏好性的,而物种在进化中会形成特有的密码子使用模式,不同物种间的密码子偏好性不同,而近缘物种间的密码子使用模式有可能相似[16]。这种偏好性的差异对揭示物种间的进化关系,探究基因特征和蛋白质功能有重要意义。本研究中,基因的密码子偏好性较弱,同义密码子的使用倾向平均,其中有19条序列密码子没有使用偏好,说明的表达水平可能相对较低,非高表达基因。统计表明碱基组成整体偏向A/T,密码子第三位也以A/U为主,这与叶绿体基因组中密码子大多偏好A/U结尾这一特征相符合[17],且GC3大多高于GC1和GC2。研究结果中94种蕨类的基因ENC值从34到61不等,其密码子偏好性存在一定差异。通常,在没有其他压力作用下,同义密码子的使用概率可能因为基因长度的限制而造成统计上的误差,这种物种间的差异性可能与之相关。同时,GC3与GC12表现出显著的相关性,密码子不同位置下碱基组成的差异较小。而ENC与GC各项数值都不相关, 说明碱基组成对密码子偏好性的影响较小。通过GC3-GC12和ENC-plot分析得出结论,基因的密码子偏好性主要受突变影响,同时也受到选择压力和基因长度等其他因素的作用。
基于凤尾蕨科和其他蕨类中基因的结构特征存在区别,对两者的分子替换速率进行比较, 研究表明,凤尾蕨科除了同义替换率外的进化速率均值都大于其他蕨类,且颠换率、非同义替换率和值间两者存在显著差异。凤尾蕨科具有陆生、水生、旱生和附生等多个生态位[18],有可能是生境的差异使在某一生态位上受到较高的选择压力,导致凤尾蕨科的进化速率升高。
在基因的适应性进化分析中,分支模型下似然比检验支持的自由比率模型中检测出15条>1的分支,说明该部分分支可能有正选择存在,其中有7条分支属于凤尾蕨科,其余分支包括了蹄盖蕨科、鳞毛蕨科、水龙骨科和合囊蕨科。同时似然比检验表示二比率模型优于单比率模型,凤尾蕨科和其他蕨类的值存在极显著差异,这与HyPhy的分子进化速率计算结果一致。但是前景支值(= 0.578 8)并不高于1,尚不能认为它是受正选择作用的,选择压力约束放松可能是较为合理的解释。基因共编码80个氨基酸位点,位点模型M2a和M8均检测出一个正选择位点74A,占总序列1.25%; 基于M8模型检测负选择位点63个(<0.05), 占总序列78.75%。当一个基因受到正选择压力时,蛋白功能可能发生改变来适应外界环境变化;而当一个基因受到负选择压力,则说明该基因倾向保持原有的功能[19]。总的来说基因大部分位点受到负选择作用,推断其进化高度保守,在蕨类中基因的结构和功能基本趋于稳定。下一步分析氨基酸位点在蛋白质结构域中的分布,对正选择位点的定位可以探究基因功能,进一步了解适应性进化发生的原因和作用。
本研究以94种蕨类植物的基因为对象,揭示了基因的基本结构特征,为叶绿体基因组的基因组成补充信息。在探讨的碱基组成与密码子偏好性的研究中,基因的密码子偏好较弱,偏好使用A和U结尾的密码子,其密码子偏好性主要受到突变的影响,并且不同物种之间密码子偏好性具有一定差异。同时,通过对基因的进化分析,发现该基因表现高度保守,为基因的功能研究提供参考。
[1] RAUBESON L A, JANSEN R K. Chloroplast genomes of plants [M]// HENRY R. Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants. Cambridge: CABI, 2005: 45–68. doi: 10. 079/9780851999043.0045.
[2] JANSEN R K, RUHLMAN T A. Plastid genomes of seed plants [M]// Genomics of Chloroplasts and Mitochondria: Advances in Photosyn- thesis and Respiration. New York: Springer, 2012: 103–126. doi: 10. 1007/978-94-007-2920-9_5.
[3] ZHANG X C, WEI R, LIU H M, et al. Phylogeny and classification of the extant lycophytes and ferns from China [J]. Chin Bull Bot, 2013, 48(2): 119–137. [张宪春, 卫然, 刘红梅, 等. 中国现代石松类和蕨类的系统发育与分类系统[J]. 植物学报, 2013, 48(2): 119–137. doi: 10.3724/SP.J.1259.2013.00119.]
[4] PPG Ⅰ. A community-derived classification for extant lycophytes and ferns [J]. J Syst Evol, 2016, 54(6): 563–603. doi: 10.1111/jse.12229.
[5] WOLF P G, ROPER J M, DUFFY A M. The evolution of chloroplast genome structure in ferns [J]. Genome, 2010, 53(9): 731–738. doi: 10. 1139/G10-061.
[6] SONG M, KUO L Y, HUIET L, et al. A novel chloroplast gene reported for flagellate plants [J]. Am J Bot, 2018, 105(1): 117–121. doi: 10. 1002/ajb2.1010.
[7] CULLIS C A, VORSTER B J, VAN DER VYVER C, et al. Transfer of genetic material between the chloroplast and nucleus: How is it related to stress in plants? [J]. Ann Bot, 2009, 103(4): 625–633. doi: 10.1093/ aob/mcn173.
[8] HSU P Y, BENFEY P N. Small but mighty: Functional peptides encoded by small ORFs in plants [J]. Proteomics, 2018, 18(10): 1700038. doi: 10.1002/pmic.201700038.
[9] KEARSE M, MOIR R, WILSON A, et al. Geneious Basic: An integrated and extendable desktop software platform for the organi- zation and analysis of sequence data [J]. Bioinformatics, 2012, 28(12): 1647–1649. doi: 10.1093/bioinformatics/bts199.
[10] ZHANG D, GAO F L, JAKOVLIĆ I, et al. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies [J]. Mol Ecol Resour, 2020, 20(1): 348–355. doi: 10.1111/1755-0998.13096.
[11] SUEOKA O. Total cross section measurements for positron and electron scattering on benzene molecules [J]. J Phys B: At Mol Opt Phys, 1988, 21(20): L631. doi: 10.1088/0953-4075/21/20/003.
[12] WRIGHT F. The ‘effective number of codons’ used in a gene [J]. Gene, 1990, 87(1): 23–29. doi: 10.1016/0378-1119(90)90491-9.
[13] POND S L K, FROST S D W, MUSE S V. HyPhy: Hypothesis testing using phylogenies [J]. Bioinformatics, 2005, 21(5): 676–679. doi: 10. 1093/bioinformatics/bti079.
[14] YANG Z H. PAML 4: Phylogenetic analysis by maximum likelihood [J]. Mol Biol Evol, 2007, 24(8): 1586–1591. doi: 10.1093/molbev/msm088.
[15] JIANG Y, DENG F, WANG H L, et al. An extensive analysis on the global codon usage pattern of baculoviruses [J]. Arch Virol, 2008, 153 (12): 2273–2282. doi: 10.1007/s00705-008-0260-1.
[16] ROMERO H, ZAVALA A, MUSTO H. Codon usage inis the result of strand-specific mutational biases and a complex pattern of selective forces [J]. Nucl Acids Res, 2000, 28(10): 2084–2090. doi: 10.1093/nar/28.10.2084.
[17] ZHOU M, LONG W, LI X. Patterns of synonymous codon usage bias in chloroplast genomes of seed plants [J]. For Stud China, 2008, 10(4): 235–242. doi: 10.1007/s11632-008-0047-1.
[18] SCHUETTPELZ E, SCHNEIDER H, HUIET L, et al. A molecular phylogeny of the fern family Pteridaceae: Assessing overall relation- ships and the affinities of previously unsampled genera [J]. Mol Phylo- genet Evol, 2007, 44(3): 1172–1185. doi: 10.1016/j.ympev.2007.04.011.
[19] PRINCE V E, PICKETT F B. Splitting pairs: The diverging fates of duplicated genes [J]. Nat Rev Genet, 2002, 3(11): 827–837. doi: 10. 1038/nrg928.
附录1 蕨类物种信息
附录2 94种蕨类植物的系统发育树
附录3基因的密码子碱基组成与ENC值
附录4基因分子进化速率
http://jtsb.ijournals.cn/ajax/common/download_attache_file.aspx?seq_id=20230218234211003&file_no
Molecular Evolution of Chloroplast Genein Ferns
LI Zifei1, SU Yingjuan2,3*, WANG Ting1*
(1. College of Life Sciences, South China Agricultural University, Guangzhou 510642, China; 2. School of Life Sciences, Sun Yat-Sen University,Guangzhou 510275, China; 3. Research Institute of Sun Yat-Sen University in Shenzhen, Shenzhen 518057, Guangdong, China)
As a newly discovered chloroplast gene in recent years,gene, with length of about 200 bp, is conserved among ferns. However, the origin and function ofrequire further investigation. The study was performed on thegene sequence of 94 species of ferns, in the phylogenetic background, analyzing codon usage bias, evolution rate and selection pressure. Results showed that the codon bias was weak ingene which the 3rd position of codon prefers A/U, variously in closely species. The codon bias was mainly produced by gene mutation. In addition, based on the differences in the structural characteristics ofbetween Pteridaceae and other ferns, their evolution rate was compared, suggesting that there were significant difference in transversion rate, nonsynonymous substitution rate and omega. Besides, only one position selection site 74A was detected. The strong negative selection pressure indicated that the function and structure ofwere mostly stabilized. The results provide a new clue of phylogenetic analysis in ferns and functional studies ofgene.
Fern;gene; Codon usage bias; Evolutionary rate; Adaptive evolution
10.11926/jtsb.4780
2023-02-18
2023-03-13
国家自然科学基金项目(31770587)资助
This work was supported by the National Natural Science Foundation of China (Grant No. 31770587).
李子菲(1998年生),女,硕士研究生,研究方向为植物系统发育与分子进化。E-mail: fuyjun420@qq.com
通讯作者 Corresponding author.E-mail: suyj@mail.sysu.edu.cn; tingwang@scau.edu.cn
猜你喜欢
东坡赤壁诗词(2023年1期)2023-05-30
生物学通报(2020年11期)2020-10-22
中成药(2019年12期)2020-01-04
疯狂英语·初中天地(2019年4期)2019-10-17
中成药(2018年7期)2018-08-04
中国民族医药杂志(2016年2期)2016-05-14
中国民族医药杂志(2016年2期)2016-05-14
中国民族医药杂志(2016年3期)2016-05-09
中国民族医药杂志(2016年3期)2016-05-09
丝绸(2015年7期)2015-02-28
