
Ⅰ型自身免疫性胰腺炎发病机制及研究进展
2023-07-31 12:47刘屹霄杨莹韵杨爱明
协和医学杂志 2023年4期
刘屹霄,杨莹韵,杨爱明
中国医学科学院北京协和医院消化内科,北京 100730
自身免疫性胰腺炎(autoimmune pancreatitis,AIP)是一种罕见的胰腺炎,患病率约10.1/10万,也称非酒精性破坏性胰腺炎和硬化性胰腺炎,属于慢性炎症性疾病[1]。组织学上以胰腺慢性炎症为特征,常表现为淋巴浆细胞浸润和纤维化。影像学可见胰腺肿大、胰管不规则狭窄。此外,AIP还是一种系统性自身免疫性疾病,可与其他自身免疫性疾病并存,也可作为IgG4相关性疾病(IgG4-related disease,IgG4-RD)谱系的一部分,在临床上体现为胰腺外组织器官受累,常见于胆管、淋巴结、肾、甲状腺等[1]。
2011年,国际胰腺病协会依据影像学、血清学、多器官受累情况、组织学和对类固醇治疗反应的不同,将AIP分为Ⅰ型和Ⅱ型[2]。Ⅰ型AIP又称淋巴浆细胞硬化性胰腺炎,Ⅱ型AIP又称特发性导管中心性胰腺炎。Ⅰ型与Ⅱ型AIP的典型区别在于Ⅰ型患者体内IgG4浓度显著增高,而Ⅱ型则不明显。目前,Ⅰ型AIP的发病机制尚未明确,为精准治疗带来了困难,但近年来其相关基础及临床研究取得了较大进展。基于此,本文将从免疫学角度阐述Ⅰ型AIP的发病机制及研究进展。
1 Ⅰ型AIP的诊断及治疗
Ⅰ型AIP的临床表现可从完全无症状至涉及多种器官受累,老年男性的无痛性梗阻性黄疸和非特异性症状(如恶心、呕吐或体质量减轻)是Ⅰ型AIP的典型症状[3]。80%的Ⅰ型AIP 患者存在IgG4相关性胆管炎[3]。无痛性黄疸可继发于胆道梗阻,而体质量减轻是胰腺功能障碍导致吸收不良的结果[4-6]。AIP将影响胰腺的内分泌和外分泌功能,导致粪便弹性蛋白酶水平降低和空腹血糖水平升高[7]。
Ⅰ型AIP的诊断可综合腹痛和黄疸等临床症状、血清IgG4变化、CT和MRI等影像学及内镜检查(如逆行性胰胆管造影等)综合判断[8]。组织病理学作为Ⅰ型AIP诊断的金标准,其典型特征为镜下可见明显的淋巴细胞浸润和组织纤维化[9]。
根据2022年发布的AIP治疗共识,AIP(包括IgG4-RD)的一线治疗药物为糖皮质激素[10]。但糖皮质激素存在一定副作用,且减量后疾病易复发。硫唑嘌呤、环磷酰胺、吗替麦考酚酯等免疫抑制剂可用于其维持治疗。近期临床上采用利妥昔单抗作为AIP的诱导和维持治疗药物,表现出良好的应用前景[11]。
2 Ⅰ型AIP的发病机制
目前文献报道的Ⅰ型AIP发病机制如图1所示。其中以适应性免疫系统T淋巴细胞-B淋巴细胞-浆细胞构成机制的主体,导致血清中IgG4升高和组织纤维化。此外,包括M2巨噬细胞、浆细胞样树突状细胞(plasmacytoid dendritic cells,pDC)、单核细胞、嗜碱性粒细胞在内的固有免疫系统也参与信号转导过程。
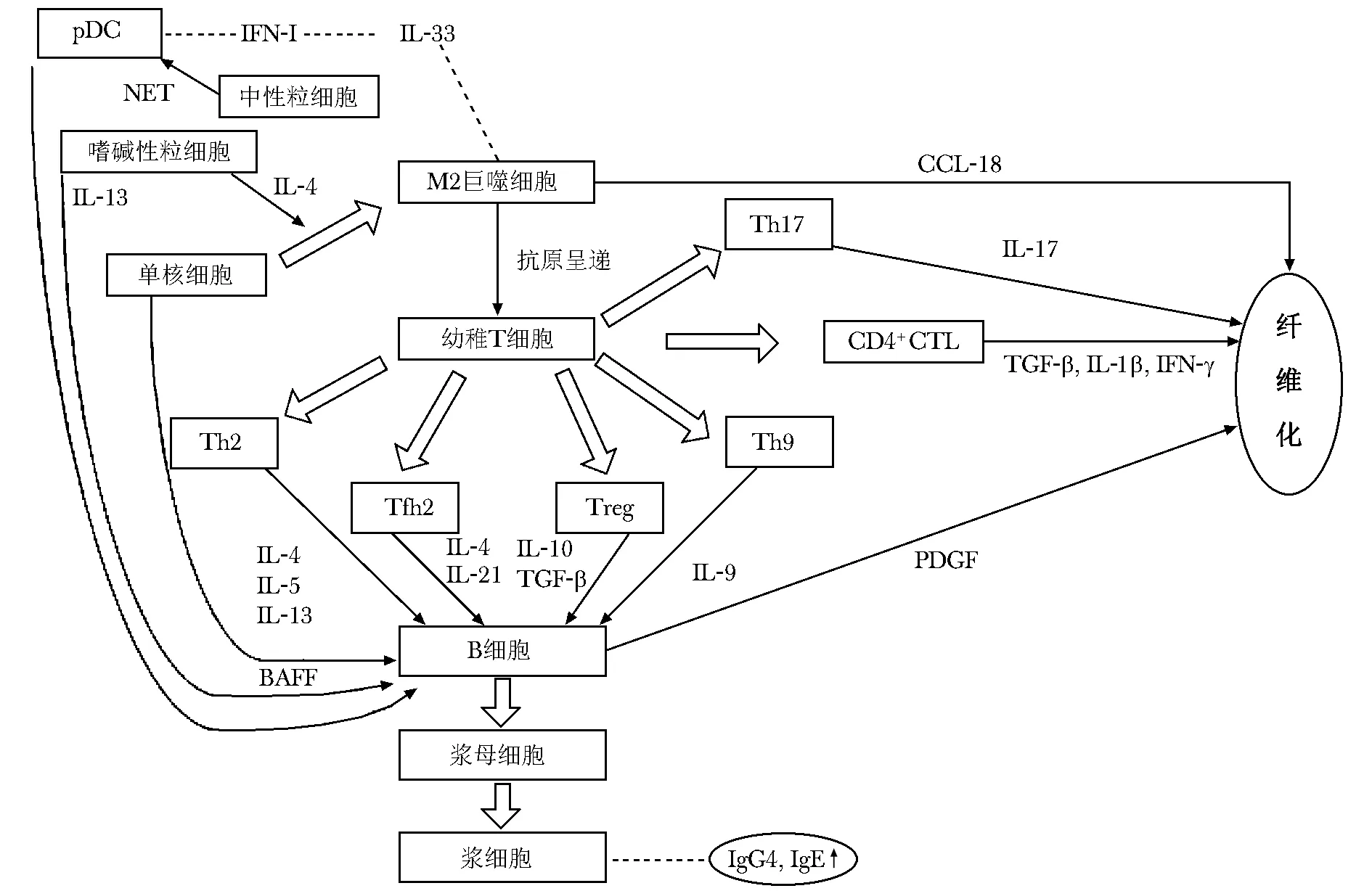
图1 Ⅰ型自身免疫性胰腺炎发病机制
2.1 适应性免疫系统
2.1.1 IgG4
Ⅰ型AIP是IgG4-RD的部分典型表现,以血清IgG4含量显著升高为主要特点。IgG4在Ⅰ型AIP发病机制中的作用尚存争议,但目前主要存在两种观点。第一种观点认为,由于IgG4对补体系统C1q和抗体Fc受体的相对弱结合而具有抗炎作用,成为一种保护因素[12]。研究发现,IgG4可与细胞膜联蛋白A11结合,从而阻止IgG1与A11的免疫复合物形成,但IgG4惰性较大,不能通过激活补体启动免疫反应,从而达到保护效果[13]。第二种观点认为,IgG4可能是一种致病因素。研究发现,将AIP患者体内的IgG1和IgG4分离,并注射至小鼠体内均体现出致病性[14]。另有观点认为,由于36%的Ⅰ型AIP患者血清IgG4含量正常,提示IgG4并未参与发病机制,仅作为疾病表象存在[15]。因此,IgG4在Ⅰ型AIP中的作用尚不明晰,难以针对其开发靶向药物,仅作为临床诊断的参考指标。
2.1.2 B淋巴细胞
B淋巴细胞可能直接参与Ⅰ型AIP的致病过程(如纤维化过程)。通过将IgG4-RD患者的幼稚B淋巴细胞、CD19+B淋巴细胞、记忆B淋巴细胞和浆母细胞与成纤维细胞共培养及免疫荧光标记,发现B淋巴细胞可通过分泌血小板衍生生长因子(platelet derived growth factor,PDGF)促进成纤维细胞的胶原合成,参与纤维化进程[16]。进一步研究发现,在Ⅰ型AIP患者中,外周血IgG4+的寡克隆B淋巴细胞含量也显著升高,并随糖皮质激素的治疗而下降,提示寡克隆B淋巴细胞在IgG4-RD患者中可能同样具有直接致病作用[17]。因此,有研究者认为IgG4-RD患者的循环浆母细胞异常升高可作为更具特异性的生物标志物[15]。
调节性B淋巴细胞(regulatory B cells,Bregs)也被发现参与了Ⅰ型AIP的调控过程。研究显示,白细胞分化抗原CD19+CD24highCD38high的Bregs在Ⅰ型AIP患者体内显著升高,而CD19+CD24highCD27+的Bregs则在患者体内出现下降趋势[18]。进一步研究发现,CD19+CD24highCD38high的Bregs在发病过程中主动增加以抑制疾病进展,同时CD19+CD24highCD27+的Bregs亦参与了疾病进展过程,但具体机制尚不明确[18]。
总而言之,B淋巴细胞广泛参与Ⅰ型AIP的发病机制,除分泌包括IgG4在内的抗体外,其信号转导功能亦不容忽视。如Bregs可通过分泌趋化因子调控疾病进展,对B淋巴细胞相关CD蛋白表达的分析有助于Ⅰ型AIP的预防和诊断。
2.1.3 T淋巴细胞
(1)辅助性T淋巴细胞(T helper cell,Th)2以分泌白细胞介素(interleukin,IL)-4、IL-5、IL-13为特征,广泛参与变态反应、超敏反应、IgE和IgG的分型转换。大量研究表明,IgG4-RD患者体内存在升高的Th2及其分泌的细胞因子(IL-4、IL-5及IL-13)[19-20],因此Th2也被认为是参与Ⅰ型AIP发病机制的关键细胞[21]。但近期有研究显示,Th2是Ⅰ型AIP患者继发其他变态反应的结果,IL-4升高可能为其他细胞(如滤泡辅助性T淋巴细胞)所分泌[22]。
(2)CD4+细胞毒性T淋巴细胞(CD4+cytotoxic T lymphocyte,CD4+CTL)被认为在Ⅰ型AIP纤维化过程中发挥关键作用。CD4+CTL分泌多种炎症因子,包括转化生长因子(transforming growth factor,TGF)-β、IL-1β、干扰素(interferon,IFN)-γ等,可激活成纤维细胞,是其参与纤维化的直接机制[23]。其他潜在机制包括表达Ⅱ类主要组织相容性复合体的细胞被CD4+CTL攻击并杀死,死亡细胞所留下的空间被激活的成纤维细胞填补,从而出现受累组织纤维化[24]。
(3)调节性T淋巴细胞(regulatory T cells,Tregs)是分泌IL-10和TGF-β等细胞因子以维持体内免疫耐受的CD4+CD25+T淋巴细胞[25]。根据来源可分为胸腺来源的FOXP+自然Tregs和外周产生的诱导性Tregs(induced regulatory T cells,iTregs)。研究发现,Ⅰ型AIP患者体内存在Tregs对受累组织的浸润,外周血中Tregs含量升高,同时循环中CD4+CD25+Treg和记忆性Tregs升高,且幼稚Tregs含量下降[26-28]。提示Tregs可影响IgG4的产生及病程进展。研究表明,诱导性共刺激分子(inducible costimulator,ICOS)阳性的Tregs可通过分泌IL-10影响B淋巴细胞,进而影响IgG4的分泌;同时ICOS阴性的Tregs则分泌TGF-β参与纤维化进程[26]。
(4)滤泡辅助性T淋巴细胞亚群抗原活化后特征性高表达C-X-C趋化因子受体5(C-X-C chemokine receptor type 5,CXCR5),后者在CXC趋化因子13的作用下使细胞定位于淋巴滤泡生发中心,进而辅助B淋巴细胞增殖、分化、产生抗体、参与体液免疫。在IgG4-RD患者的IgG4+浆母细胞的寡克隆扩增中,可见免疫球蛋白重链可变区和框架区内显著增强的重排和超突变[29],说明B淋巴细胞的扩增依赖于滤泡辅助性T淋巴细胞(follicular helper T cell,Tfh)在生发中心内的反复突变和阳性选择。事实上也观察到IgG4-RD患者体内存在大量Tfh细胞,其在促进浆母细胞和浆细胞分化的过程中发挥重要作用[30]。目前认为Tfh通过分泌IL-4和IL-21参与Ⅰ型AIP的发病过程,分泌IL-4的Tfh参与抗体类别转换,分泌IL-21的Tfh参与B淋巴细胞超突变过程[30-31]。
此外,其他T淋巴细胞也参与Ⅰ型AIP的发病机制。最新研究发现,IL-35的表达水平和Th9在IgG4-RD患者体内显著增高,Th9分泌的IL-9促进了浆细胞分化和抗体类别转换[32]。研究显示,IgG4-RD患者循环中Th17显著增高[33],Th17通过分泌IL-17参与TGF-β途径,调节成纤维细胞活动,进而调节组织纤维化,并表达转录因子RORγt,以招募中性粒细胞和巨噬细胞参与自身免疫性炎症反应[34]。
综上,T淋巴细胞群主要通过分泌多种IL和炎症因子以激活B淋巴细胞、成纤维细胞,促进浆细胞分化、抗体分泌和组织纤维化进程。临床上,作为一种上游调节机制,对T淋巴细胞分泌因子的拮抗治疗可能使患者受益,但尚需更多研究深入探索。
2.2 固有免疫系统
2.2.1 pDC
pDC是以分泌IFN-Ⅰ为特征的树突状细胞[35]。pDC表达Toll样受体(Toll-like receptor,TLR)7和TLR9,其接收到来自细菌或病毒的单链RNA或双链DNA后将启动下游的若干蛋白,包括Myd88、IRAK4等,最终形成包含干扰素调节因子7(interferon regulatory factor,IRF7)的复合体,而IRF7可直接调控IFN-Ⅰ的表达[36-39]。值得注意的是,IFN-Ⅰ的表达正反馈作用于细胞表面的IFN-Ⅰ受体,促进IRF7的表达,进一步增加IFN-Ⅰ的分泌,这种正反馈机制导致相关自身免疫性疾病的发生[38,40]。
研究表明,pDC参与了Ⅰ型AIP与IgG4-RD的发病机制,其通过pDC-IFN-Ⅰ-IL-33轴实现对炎症过程的调控[41]。pDC通路的启动可能来自于中性粒细胞胞外杀菌网络(neutrophil extracellular trap,NET)和肠道菌群失调[42-43]。启动pDC上的TLR后,IFN-Ⅰ分泌增加,进一步促进IL-33的分泌,IL-33可诱导胰腺炎慢性纤维化炎症反应[41,44]。将pDC耗竭及中和阻断IFN-Ⅰ-IL-33通路可有效抑制实验模型AIP的发展,提示此信号通路作为AIP潜在的治疗靶点具有可行性[45]。此外,pDC还可通过分泌B淋巴细胞激活因子(B-cell activating factor,BAFF)诱导B淋巴细胞分泌IgG4[42,46]。pDC作为天然的免疫细胞,在Ⅰ型AIP发病机制中占据上游,通过启动pDC-IFN-Ⅰ-IL-33轴以及分泌BAFF参与通路激活,以pDC为治疗切入点的pDC耗竭疗法及通路阻断可提高AIP的治愈率,并降低其复发率。
2.2.2 巨噬细胞
AIP患者胰腺组织内可见M2巨噬细胞浸润[47]。类似于上文所述的pDC-IFN-Ⅰ-IL-33轴,M2巨噬细胞上的TLR7同样可被外界病毒或内源性RNA激活,促使细胞分泌IL-33,导致炎症和纤维化[48-49]。此外,分泌趋化因子CCL(C-C motif chemokine ligand,CCL)-18的M2巨噬细胞被发现具有重要作用。CCL-18具有参与胶原蛋白合成的功能,研究发现CCL-18在Ⅰ型AIP患者体内高表达,可能参与了IgG4-RD的组织纤维化进程,提示可将CCL-18作为表征疾病进程和严重程度的生物标志物[50-51]。针对以巨噬细胞为中心的信号通路进行检测和拮抗可能为临床诊疗Ⅰ型AIP的新思路,但仍需更多相关基础研究进行验证。
2.2.3 嗜碱性粒细胞
在自身免疫性疾病进展过程中,嗜碱性粒细胞将产生细胞因子并作为抗原呈递细胞参与免疫反应[52]。研究表明,在外源性抗原(如微生物)的刺激下,嗜碱性粒细胞上的TLR被激活,可促进IL-13和BAFF的分泌,作用于B淋巴细胞,增强IgG4的分泌,从而参与疾病进展[53-54]。此外,嗜碱性粒细胞也可通过分泌IL-4介导单核细胞向M2巨噬细胞转化,间接参与Ⅰ型AIP的组织纤维化过程[55]。
2.2.4 自身抗原
尽管疾病的特异性靶点尚未明晰,但IgG4-RD是一种全身受累性疾病,提示所涉及器官中存在共同靶抗原的可能。对于Ⅰ型AIP,自20世纪90年代以来,已经发现碳酸酐酶、纤溶酶原结合蛋白、乳铁蛋白、胰腺分泌胰蛋白酶抑制剂、淀粉酶、胰蛋白酶原、膜联蛋白A11、层粘连蛋白511、半乳糖凝集素3等自身抗原在其中发挥作用[13,47,56-57]。这些自身抗原对应的抗体在Ⅰ型AIP患者体内异常增高,参与了器官损伤和发病过程。众多自身抗原的发现解释了IgG4-RD全身受累的原因,也提示可早期通过对自身抗原相关基因的筛查发现Ⅰ型AIP发病的可能性,在临床上及时进行有针对性的评估和检查。
3 小结与展望
综上所述,免疫失调是导致Ⅰ型AIP组织浸润、炎症和纤维化等临床症状的重要途径。以B淋巴细胞、浆母细胞为代表的体液免疫系统参与了Ⅰ型AIP的血清学症状改变,如IgG4升高;包括M2巨噬细胞、T淋巴细胞家族、B淋巴细胞在内的广泛免疫系统通过不同途径促进Ⅰ型AIP的组织纤维化进程;pDC、嗜碱性粒细胞等则在信号传导过程中发挥重要作用。虽然已证明免疫系统在Ⅰ型AIP发病机制中的广泛参与,但相关基础研究主要聚焦于单个细胞或分子的作用,而Ⅰ型AIP中代谢网络和信号转导网络尤其是各信号通路间的相互作用机制尚不明晰,仍需更多研究深入探索。此外,目前Ⅰ型AIP的激素类一线治疗药物存在副作用大、易复发等缺点,期待未来进一步揭示其发病机制,并针对Ⅰ型AIP免疫信号通路进行靶向药物开发,以使患者更大化受益。
作者贡献:刘屹霄和杨莹韵负责查阅文献、撰写及修订论文;杨爱明负责设计选题和审校论文。
利益冲突:所有作者均声明不存在利益冲突
猜你喜欢
中国民间疗法(2021年13期)2021-08-30
中国医学影像学杂志(2021年6期)2021-08-13
天津医科大学学报(2021年2期)2021-03-29
现代临床医学(2019年6期)2019-12-07
中国生殖健康(2019年2期)2019-08-23
肝胆胰外科杂志(2015年4期)2015-02-27
西南军医(2014年5期)2014-04-25
中医研究(2014年5期)2014-03-11
西南军医(2014年1期)2014-02-03
中医研究(2013年5期)2013-03-11
