
基于热还原氧化石墨烯的单分子表面诱导荧光衰逝技术*
2023-07-27 10:59:56樊秦凯杨晨光胡书新徐春华李明陆颖
物理学报 2023年14期
樊秦凯 杨晨光 胡书新 徐春华 李明 陆颖†
1) (中国科学院物理研究所,北京凝聚态物理国家研究中心,软物质物理重点实验室,北京 100190)
2) (中国科学院大学,北京 100049)
单分子表面诱导荧光衰逝(single molecule surface-induced fluorescence attenuation,smSIFA)技术是一种基于二维材料受体、用于研究生物大分子法向运动的精密测量方法,该方法不受二维平面运动的干扰.作为受体的二维材料,其特征淬灭距离决定法向上探测的距离和精度.近年来以氧化石墨烯(graphene oxide,GO)和石墨烯作为介质受体的SIFA 技术在生物大分子的研究中发挥了重要作用,但石墨烯和GO 具有固定的特征淬灭距离,探测范围有限.调整探测范围需要更换介质材料,面临材料选择与制备的困难,亟需开发用于技术的可调控材料.本文改良了以GO 为介质受体的单分子SIFA 技术,利用热还原的方法对GO 进行还原,通过控制还原温度,制备出了还原程度不同的还原氧化石墨烯(reduced graphene oxide,rGO),调控特征淬灭距离,利用荧光标记的DNA 测量rGO 的特征淬灭距离.将rGO 用于单分子SIFA 技术,对Holliday junction 构象变化的观察,论证了rGO 的探测范围.
1 引言
单分子荧光共振能量转移(single molecule fluorescence resonance energy transfer,smFRET)是研究生物大分子动力学过程的常用方法,具有较高的时间和空间分辨率[1-4].膜蛋白是生物膜功能的主要承担者,在细胞内约有1/3 的基因编码膜蛋白[5].研究膜蛋白在细胞膜上的取向和插膜深度对于理解其结构与功能至关重要[6,7].由于细胞膜具有流动性,处于细胞膜上的膜蛋白会发生横向位移,将FRET 用于膜蛋白的研究时具有局限性[8-10].近年来基于荧光共振能量转移原理发展的表面诱导荧光衰逝(surface-induced fluorescence attenuation,SIFA)技术是将二维介质作为荧光受体,通过分析其对荧光供体的光强衰逝程度,可以计算出荧光供体距和材料表面的法向距离[11,12].荧光衰逝效率和供体距介质表面法向距离的关系由(1)式描述:
式中:I0为没有介质存在时荧光供体的初始强度;I为当介质存在时测得的供体荧光强度;d0为介质的特征淬灭距离,是I/I0=0.5 时荧光供体和介质表面的法向距离.在选择二维介质作为荧光受体时不仅要考虑到材料的亲生物性,同时还要考虑到材料的光学性能以及制备难易度.近年来基于氧化石墨烯(graphene oxide,GO)的SIFA 方法在膜蛋白的研究中有着广泛的应用[13,14],基于石墨烯的单分子荧光成像也取得了一定成果[15,16].以往研究表明,GO 的d0=4 nm[11],石墨烯的d0≈ 18 nm[16-18].GO 的d0较小,适合探测距离其表面2—6 nm 范围内的信号[11];石墨烯的d0较大,适合探测距离其表面12 nm 以外的区域[16].图1(a)展示了利用GO和石墨烯作为介质受体时的局限,以石墨烯和GO作为介质受体时并不能全方位的覆盖近细胞膜表面的区域,存在无法探测的区间.图1(b)展示了不同d0的距离淬灭曲线,在d0附近衰减曲线的斜率最大,探测的灵敏度最高,当荧光供体距离介质表面较近或较远时探测的灵敏度显著下降.不同的二维材料的d0不同,能够探测的范围不同,进行SIFA实验时需要根据目标大分子的尺寸以及荧光标记位点等信息来选择具有目标d0的材料.

图1 调控d0 的SIFA 方法 (a) SIFA 方法示意图;(b) 荧光供体光强衰减和表面距离的关系;(c) SIFA 探测灵敏度和荧光供体距表面距离的关系Fig.1.SIFA method of adjustable d0: (a) Schematic representation of SIFA method;(b) relationship between degree of attenuation of a fluorescent donor and donor-surface distance;(c) relationship between detection sensitivity of SIFA and donor-surface distance.
SIFA 方法探测荧光供体距离细胞膜法向距离的变化是通过测量供体的光强来实现的,因此SIFA的灵敏度受制于仪器对于荧光供体光强的探测.通常认为仪器能够分辨的光强变化率约为10%[11],基于此可以利用(1)式计算出不同d0的距离探测灵敏度.图1(c)展示了SIFA 探测的距离灵敏度曲线,GO 的d0=4 nm,在d0附近的探测灵敏度可以接近0.5 nm,在距离其表面2—6 nm 的范围内可以实现1 nm 的分辨率,然而一旦超出3—6 nm 的灵敏范围之后,GO 的探测分辨率将低至3 nm,并且随着距离的增加分辨率呈指数下降.当d0提高到10 nm 时,虽然能够探测距离介质表面距离更远范围的荧光信号,但即便是在最灵敏的d0附近也只能实现1 nm 的探测精度.在进行SIFA 实验时,如果要实现1 nm 的空间分辨率,则介质受体的d0应不大于10 nm.如图1(c)所示,要对介质受体表面12 nm 以内的生物大分子进行探测,以弥补石墨烯和GO 探测的局限,并保证1 nm 的空间分辨率,需要对d0在4—10 nm 之间进行连续调节.制备d0在3—10 nm 的介质材料并用于SIFA实验来观察生物大分子面临材料选择和制备的困难.有研究表明,对石墨烯进行通电使其氧化可以减小d0,但石墨烯氧化具有不均匀性[19].还原氧化石墨烯(reduced graphene oxide,rGO)是GO 通过加热、化学和电学等方法进行还原后所得到的材料,rGO制备方便,且物理和化学性质和石墨烯接近[20-22].有研究表明,rGO 对荧光的淬灭效果介乎与GO 和石墨烯之间[23,24],但rGO 仍缺乏单分子荧光成像的应用.本文采用热还原方法制备rGO,并用于单分子荧光成像.通过荧光白标记的双链DNA 标尺精确测量了不同还原程度rGO 的d0,改良了已有的SIFA 技术,并通过与GO进行对比,验证了rGO-SIFA 技术的优势.
2 实验材料与方法
2.1 GO 分散液的制备
GO 分散液制作方法参照 Hummers 方法[25,26].取1 g NaNO3,46 mL H2SO4以及1 g 石墨,于0 ℃温度下混合均匀,对混合物进行冰浴以保持其环境温度为0 ℃,并向混合物中缓慢加入6 g KMnO4.此后不断搅拌约2 h,而后将混合物整体移至35 ℃继续搅拌2 h,将120 mL 去离子水缓慢加入混合溶液中(30 min),最后将6 mL H2O2(质量分数为30%)缓慢加入混合溶液中,并培养20 min 左右.超高速离心(15000 r/min) 20 min 制得的混合溶液,去除上清液,将得到的沉淀再次分散溶于去离子水中,进行超声(100 W,30 min),对多层氧化石墨进行剥离.对溶液进行低速离心(1000 r/min)10 min,去除沉淀保留上清液,重复该步骤3—4 次,直到不再出现明显的沉淀.将收集到的溶液进一步离心,每次离心取走上清液后将沉淀再次溶解进行下一次离心.分别以8000,6000,4000 r/min离心25 min,4000 r/min 离心后的沉淀溶解后再次以2000 r/min 离心25 min,所得到的上清液即为实验所用的GO 分散液.
2.2 XPS 方法分析GO 的还原
用硝酸纤维素膜(直径47 mm,孔径0.2 mm,Whatman)对制备的GO 分散液进行真空抽滤[27],所形成的薄膜自然晾干之后从滤膜上分离下来.剪取少量GO 薄膜,用2 片干净的盖玻片将GO 薄膜夹在中间,放置于真空管式炉内烘烤,升温速度设为8 ℃/ min,升温至目标温度后维持2 h,而后自然冷却至室温.将加热还原后的rGO 薄膜与GO薄膜在中国科学院物理研究所的X 射线光电子能谱仪(XPS,Thermo Fisher Scientific ESCALAB 250X)上测量结合能,所用X-射线源为单色化的铝Kα射线,能量为1486.6 eV.
2.3 rGO-SIFA 实验步骤
rGO-SIFA 实验包含rGO-SIFA 样品腔室制备、单分子荧光实验材料与样品制备,以及荧光观测三部分.
2.3.1 rGO-SIFA 样品腔室的制备过程
实验所用的盖玻片需要清洗掉表面的荧光杂质,以免对单分子荧光成像实验造成影响.具体清洗流程是先使用丙酮对盖玻片超声清洗30 min,然后用去离子水超声清洗洗去丙酮残留,再用甲醇超声清洗30 min,并在超声清洗结束后用去离子水超声清洗洗去甲醇残留.用1 mol/L 的NaOH溶液对盖玻片进行超声清洗,每次清洗10 min 后更换NaOH 溶液,共超声清洗3 次,得到干净且没有荧光杂质的盖玻片.
利用LB(Langmuir-Blodgett)技术将GO 分散液中的单层GO 转移至清洗干净的盖玻片上[28],将另一块干净的盖玻片覆盖其上,放置于真空管式炉内烘烤,升温速度设为8 ℃/min,升温至目标温度后维持2 h,而后自然冷却至室温.用双面胶将rGO 盖玻片和清洗干净的载玻片黏合在一起,做成适合装载在全内反射荧光显微镜上的样品腔室.
2.3.2 单分子荧光实验材料与样品制备
实验所用的DNA 序列采购于上生工生物工程(上海)股份有限公司,双链DNA 的序列为TATGGTCAACTGCTGAGCGTAG-biotin,ATACCAGTTGACGACTCGCAT.DNA Holiday junction 的4 条链序列如表2 所示.退火缓冲液(pH=7.5)成分为50 mmol/L NaCl,25 mmol/L Tris-HCl.双链DNA 退火时将2 条单链DNA 按照1∶1 混合,退火缓冲液(pH=7.5)成分为50 mmol/L NaCl,25 mmol/L Tris-HCl,加热至95 ℃孵育5 min,而后在7 h 内缓慢冷却至室温.
观察双链DNA 样品时的缓冲液成分为50 mmol/L NaCl,25 mmol/L Tris-HCl,pH=7.5.观察DNA Holiday junction 样品时的缓冲液成分为50 mmol/L NaCl,50 mmol/L MgCl2,25 mmol/L Tris-HCl,pH=7.5.在进行单分子荧光成像时缓冲液内需要加入抗淬灭体系,成分为0.8% D-葡萄糖、1 mg/mL 葡萄糖氧化酶、0.4 mg/mL 过氧化氢酶、1 mmol/L Trolox.
2.3.3 rGO-SIFA 荧光观测的步骤
首先,将1 mg/mL 生物素修饰的牛血清蛋白(biotin-BSA)注入样品腔室,孵育5 min 后用PBS缓冲液冲洗掉游离的biotin-BSA;加入10 µg/mL链霉亲和素(strepavidin,SA)溶液孵育5 min 后用PBS 缓冲液冲洗掉游离的SA.然后,将100 pmol/L末端标记了biotin 的荧光标记DNA 注入样品腔室,孵育5 min 后用PBS 缓冲液冲洗掉游离的DNA.最后,将缓冲液和抗淬灭体系混合注入样品腔内进行单分子荧光成像.实验装置采用以全内反射荧光显微镜和EMCCD 为基础的双通道荧光共振能量转移观测装置[29-31],EMCCD 曝光时间设为50 ms.利用ImageJ 和Matlab 等软件记录及分析光强数据.
3 结果与讨论
3.1 XPS 分析GO 的热还原
利用LB 技术可以便捷、低成本地在盖玻片上修饰单层GO,以制备成样品腔室进行单分子SIFA实验[11].实验所用的盖玻片最高能够在600 ℃高温下不发生断裂与形变,基于此,将修饰了单层GO的盖玻片直接放置于真空管式炉内进行烘烤,便可得到修饰了rGO 的盖玻片,并进行SIFA 实验.
不同还原程度的rGO 在物理、化学和光学等性质上可能存在区别[32].通过XPS 方法测量GO与rGO 的表面化学成分和结合状态,可以分析经过管式炉烘烤后GO 的还原情况[33].单层GO 片径大多在微米级别[11],小于XPS 的探测范围,直接对盖玻片上的rGO 进行测量时玻璃成分的盖玻片会对实验结果产生干扰,因此将GO 分散液制备成GO 薄膜,用2 块盖玻片将GO 薄膜夹在中间,与修饰了GO 的玻片同时烘烤,并对热还原后的rGO 薄膜进行XPS 分析.
如图2(a)所示,GO 的C1S 谱图展示出了3 个峰,分别对应C—C (sp2杂化碳)、C—O(羟基和环氧化物)、C=O(羰基)[27].经过300 ℃和400 ℃烘烤2 h 进行还原的rGO 其C—O 与C=O 的峰明显变低(图2(b),(c)),表明大部分含氧基团已被去除.XPS 全谱的结果也表明经过烘烤后GO 得到了很好的还原,如图2 右侧一列所示,未还原的GO,300 ℃温度下烘烤2 h 还原的rGO (300 ℃-2h-rGO),以及400 ℃温度下烘烤2 h 还原的rGO(400℃-2 h-rGO),C/O 比值分别是1.14,2.48 和3.29.这一结果和以往的研究一致,热还原rGO 的还原程度主要取决于还原温度和还原时间[34,35].还原温度和还原时间的调节是连续的,通过设定不同还原参数可以对rGO 的还原程度进行连续调控.

图2 初始GO 以及rGO 薄膜的XPS 谱 (a) 初始GO 薄膜C 1s 的XPS 谱(左)以及XPS 全谱(右);(b) 300 ℃-2 h-rGO 薄膜C 1s 的XPS 谱(左)以及XPS 全谱(右);(c) 400 ℃-2 h-rGO 薄膜C 1s 的XPS 谱(左)以及XPS 全谱Fig.2.XPS spectra of original GO and rGO thin films: (a) C 1s XPS spectra (left) and XPS survey spectra (right) for original GO thin films;(b) C 1s XPS spectra (left) and XPS survey spectra (right) for 300 ℃-2 h-rGO thin films;(c) C 1s XPS spectra (left)and XPS survey spectra (right) for the 400 ℃-2 h-rGO thin films.
3.2 热还原rGO 特征淬灭距离d0 的测定
在应用以rGO 作为介质受体的SIFA 技术研究生物大分子在细胞膜表面的法向运动之前还面临2 个问题: 一是验证rGO 是否能够用于生物大分子的研究;二是测定rGO 的d0.双链DNA 在溶液中的结构稳定,荧光标记的DNA 不仅可以作为单分子荧光成像技术的标准样品来验证方法的可行性,还可以用于研究与DNA 相互作用的蛋白质.将同时标记了Cy3 和biotin 的双链DNA 通过biotin-BSA 以及SA 连接在rGO 表面,如图3(a)所示.通过测量Cy3 的光强,与玻璃表面测得的光强进行对比,即可利用(1)式计算出rGO 的d0.图3(b)左列是在DNA 未链接到表面上之前对样品腔室进行成像,右列是DNA 连接后的成像图.经过热还原后的rGO 已不自发荧光,通过图像已无法区分荧光点处于rGO 区域还是玻璃区域,因此需要分析和统计所有单个Cy3 分子的光强.图3(c)—(e)的首行分别展示了Cy3 标记在双链DNA 第1 bp、第9 bp、第21 bp 位点时单个Cy3 分子的光强曲线,以及光强的统计图.结果表明: Cy3 标记在双链DNA 上时的光强稳定,并且荧光强度和标记位点无关.Cy3 的光强呈高斯分布,峰值即为I0.图3(c)第2 行和第3 行分别是在300 ℃-2 h-rGO和400 ℃-2 h-rGO 样品腔内对Cy3 标记在第1 bp的DNA 进行成像所得到的单个Cy3 的荧光强度曲线以及强度分布.在rGO 样品腔内观察到Cy3具有2 个稳定的光强,较高的光强和玻璃上成像的强度相同,表明此时DNA 连接在了玻璃上.较低的光强是因为DNA 连接在了rGO 上,rGO 对Cy3有衰逝作用,从而导致Cy3 光强降低.Cy3 光强较低的曲线光强稳定、不波动,表明Cy3 距离rGO的高度保持恒定.

图3 荧光标记DNA 测量rGO 的d0 (a) DNA 成像实验示意图;(b) 在300 ℃-2 h-rGO (上)以及400 ℃-2 h-rGO (下)样品腔内观察DNA;Cy3 标记在1 bp (c),9 bp (d)和21 bp (e) 处的DNA 在玻璃以及rGO 上成像时的单分子光强Fig.3.Determination of d0 of rGO by fluorescence labeled DNA: (a) Schematic representation of DNA imaging;(b) DNA imaging on 300 ℃-2 h-rGO (upper) and 400 ℃-2 h-rGO (lower);intensities of Cy3 labeled at 1 bp (c),9 bp (d) and 21 bp (e) of DNA on glass and rGO.
300 ℃-2 h-rGO 和400 ℃-2 h-rGO 样品腔内Cy3的低光强存在差异,分别是0.66I0和0.45I0,表明400 ℃-2 h-rGO 对荧光的衰逝作用比300 ℃-2 h-rGO要强.在rGO 样品腔对Cy3 标记在第9 bp 的DNA进行成像并分析单个Cy3 分子的光强,如图3(d)所示,当Cy3 标记在第9 bp 时光强比标记在第1 bp 时要强,这是由于Cy3 距离rGO 表面的距离比标记在1 bp 时要远.在300 ℃-2 h-rGO 样品腔内对Cy3 标记在第21 bp 的DNA 进行成像,此时单个Cy3 分子只有一个光强,且和玻璃样品腔内测得的光强接近,表明此时rGO 对Cy3 的荧光衰逝效果已经不显著了.统计结果表明Cy3 的光强只有一个峰,但峰宽较玻璃上测得的I0分布要宽.Cy3 标记在第21 bp 的DNA 在400 ℃-2 h-rGO样品腔内依旧能够展示出2 个光强分布,表明400℃-2 h-rGO 具有更强的荧光衰逝效果.
双链DNA 在溶液中的驻留长度为50 nm[36],相邻碱基对的距离是0.34 nm[37],因此实验所用的21 bp 双链DNA 可以近似看作是长7.1 nm 的杆状刚体.在计算Cy3 与rGO 表面的高度时只需要考虑双链DNA 与rGO 表面法向的夹角.有研究表明溶液中固定在表面的短双链DNA 和表面法向的夹角是恒定的,并且夹角约为60°[38,39],再结合以往研究表明BSA 的尺寸大约是3 nm[40],SA 的尺寸大约是4.2 nm[41],Cy3 标记在第1 bp、第9 bp以及第21 bp 时和rGO 表面的法向距离便可以计算出来.表1 总结了不同荧光标记的DNA在玻璃以及rGO 上进行成像,利用光强与距离信息计算出的d0.在相同的rGO 样品腔内对不同荧光标记的DNA 进行成像并计算d0,所得到的结果接近,更加证实了rGO-SIFA 方法的可行性.在相同还原条件的rGO 上对不同荧光标记位置的DNA 进行成像并计算d0属于独立实验,可将独立实验的结果联合起来得到更为准确的结果,通过加权平均计算后可以得到300 ℃-2 h-rGO 的d0=(6.3 ±0.5) nm,400 ℃-2 h-rGO 的d0=(7.9 ± 0.5) nm.将计算得到的300℃-2 h-rGO 的d0以及Cy3 标记在第21 bp时的高度代入(1)式可以计算出Cy3 的理论光强,大约为0.9I0.由于单分子的差异以及仪器测量存在误差,Cy3 的光强呈高斯分布,具有一定的峰宽,即便光强分布具有0.9I0和I0两个峰也无法区分,和实验测量的结果一致.300 ℃-2 h-rGO 和400 ℃-2 h-rGO 的d0存在差异,可以预计的结果是通过降低还原温度d0能从6.3 nm逐渐向4 nm 过渡.本文采用最高还原温度为400 ℃,距离实验所用盖玻片的600 ℃耐高温上限还有升温空间.另一方面也可以将玻璃盖玻片更换为石英盖玻片,从而实现更高温度的热还原,以进一步提高d0.

表1 荧光标记DNA 测量rGO 的d0Table 1. Determination of d0 of rGO by fluorescence labeled DNA.
3.3 rGO-SIFA 测量DNA Holliday junction 构象变化
将不同d0的受体介质用于单分子SIFA 技术时能够探测距离细胞膜表面不同高度的生物大分子动力学过程.利用荧光标记的双链DNA 来测量rGO 的d0时在rGO 上连接的DNA 只有一个光强,即目标生物大分子只有一个状态.为了更好地验证rGO-SIFA 方法的可行性,应选择一个荧光标记位点距离rGO 高度发生变化的生物大分子来进行研究,这个生物大分子在溶液中的结构稳定,并且被研究得比较充分.Holliday junction 是DNA复制以及同源重组的重要中间体,是由4 条DNA链组成的四链结构[42].在溶液中没有2 价金属离子存在时,Holliday junction 呈现出静止的十字形结构,当溶液中有2 价金属离子存在时,Holliday junction 将堆叠成X 型结构[43].Holliday junction堆叠成的X 型结构存在2 种不同的构象,并且这2 种构象在不断地互相转换,转换速率受到2 价金属离子浓度的调控[44,45].Holliday junction 的构象变换是发生同源重组的必要条件,其中心区域的核苷酸序列会影响2 种构象的转换速率以及构象持续时间[45,46].由于Holliday junction 的结构简单、易于制备,且在溶液中能够稳定观察到2 种构象的互相转换,荧光标记的Holliday junction 常用于高级单分子荧光成像技术的验证[47,48].构建如图3 所示的Holliday junction,表2 展示了构成Holliday junction 的4 条短链DNA 的核苷酸序列,以及Cy3的标记位置,该序列的Holliday junction 可被连接到盖玻片表面,并在2 价金属离子存在时存在稳定的构象变换[46,48].

表2 DNA Holliday junction 的核苷酸序列Table 2. Nucleotide sequence of DNA Holliday junction.
Cy3 标记在H 链的3’末端,Holliday junction通过H 链5’末端标记的biotin 固定在表面.在50 mmol/L Mg2+,50 mmol/L Na+条件下Holliday junction 不断地发生构象变化,处于state 1 时Cy3距离rGO/GO 或玻璃表面距离近(约7.5 nm),处于state 2 的时候距离表面距离远.
图4(a)是玻璃样品腔内对Holliday junction进行观察的结果,左列展示了单个Cy3 分子的光强,光强稳定未发生任何波动,统计结果表明Cy3的光强为高斯分布,中心值即为I0.图4(b)是在GO 样品腔内对Holliday junction 进行观察,未观测到Cy3 的光强发生变化.Cy3 距离表面最近的距离约为7.5 nm,GO 的d0=4 nm,通过(1)式可以计算出Cy3 距离GO 较近时的理论光强为0.94I0.距离7.5 nm 已经超出了GO 的灵敏范围,此时随着距离的增大光强从0.94I0逐渐靠近I0,这一微小的光强变化已经低于仪器探测的灵敏度.对GO 上Cy3 的光强进行统计,结果为高斯分布,但半高宽较玻璃上要大.这一现象的原因有2 个:一是理论计算表明GO 上Cy3 有2 个光强,并且这2 个光强大小都和I0很接近,实验上很难区分出这2 个光强;二是GO 本身会自发微弱的荧光,在GO 上对单分子进行成像所得到的信噪比要比玻璃上差,因此光强分布的峰宽会比玻璃上要大.图4(c)是在400 ℃-2 h-rGO 样品槽内对Holliday junction 进行观察,Cy3 的光强不断在高和低2 个值之间跳变.在400 ℃-2 h-rGO 上观察Cy3 标记的双链DNA 时Cy3 的光强稳定、不波动,表明400 ℃-2 h-rGO 只对Cy3 的光强有衰逝作用,但并不影响Cy3 光强的稳定性.因此,观察到Holliday junction所标记的Cy3 光强发生变化是由于Cy3 距离400℃-2h-rGO 的高度发生了变化.对Cy3 的光强进行时间加权统计,结果表明Cy3 的光强呈现2 个峰的分布,峰值分别是0.42I0和0.83I0.利用(1)式,代入400 ℃-2 h-rGO 的d0=(7.9±0.5) nm 可以计算出Cy3 与rGO 表面的法向距离分别是7.3 nm以及11.7 nm,与Cy3 的理论距离7.5 nm 以及11.1 nm 比较接近.根据图4(c)中Cy3 的光强分布图可以计算出高光强和低光强的峰高比为1∶2,表明Holliday junction 的state 1 构象持续时间是state 2 的2 倍,和以往采用相同DNA 序列的Holliday junction 研究结果一致[46].基于以上两点,证实了用400 ℃-2 h-rGO 观察到了Holliday junction的构象变换,并且这一构象变换无法被GO 观察到.造成这一结果差异的原因是400 ℃-2 h-rGO具有比GO 更大的d0,能够观测更远的范围.热还原的rGO 可以通过控制还原温度来调控d0,在进行具体研究时应根据目标大分子的尺度和荧光标记位点来灵活设还原温度,以选择适合观测的d0.
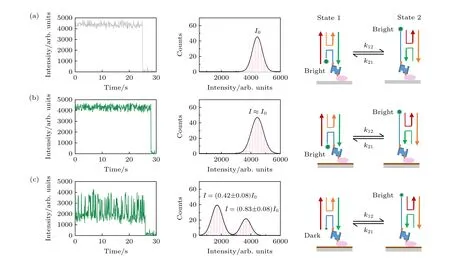
图4 SIFA 观察Holliday junction 的构象变换,在玻璃 (a),GO (b)和400 ℃-2 h-rGO (c)上观察Cy3 标记的Holliday junction,左列为单个Cy3 的光强时间曲线,中间为Cy3 的光强统计图,右列为Holliday junction 构象变换导致Cy3 光强变化的示意图Fig.4.Observing conformational transformation of Holliday junction by SIFA,observing the Cy3 labeled Holliday junction on glass(a),GO (b) and 400 ℃-2 h-rGO (c),left columns show intensity-time curves of a single Cy3,middle columns show distribution of intensities of Cy3,right columns show schematic representation of the change of Cy3 light intensity caused by the conformational transformation of Holiday junction.
4 结论
本文进一步拓展了课题组研究的SIFA 技术,将修饰了GO 的盖玻片置于真空管式炉内烘烤,通过控制温度获取不同还原程度的rGO,从而调控特征淬灭距离,使得该方法广泛用于具有二维体系的单分子研究.本文采用荧光标记的DNA 测量出了300 ℃-2 h-rGO 的d0=(6.3±0.5) nm,400 ℃-2 hrGO的d0=(7.9±0.5) nm.可预计的结果是在室温和400 ℃之间改变还原温度能够实现d0从4 nm到7.9 nm 的连续调节.本文分别在400 ℃-2 h-rGO和GO 上对Cy3 标记的Holliday junction 进行观察,GO 不能探测到Cy3 光强发生变化,而400 ℃-2 h-rGO 上可以看到Cy3 的光强不断地在高低之间跳变,并通过光强计算出了Cy3 和rGO 表面的距离,计算结果和理论距离接近,表明基于rGO的SIFA 不仅提升了SIFA 的适用范围,更具有较高的测量精度以及空间分辨率.此外,由于rGO 本身不发荧光,在进行单分子荧光成像时并不会影响图像的信噪比,将SIFA 技术和FRET 技术联用起来可以实时地观察生物大分子的三维运动与构象变换信息.期待未来在膜蛋白功能和结构的研究中,rGO-SIFA 技术能得到更广泛的应用.
猜你喜欢
昆钢科技(2022年1期)2022-04-19 11:36:14
产业用纺织品(2021年8期)2021-12-31 01:54:22
中学生物学(2021年8期)2021-11-02 04:53:14
海外星云 (2021年21期)2021-01-19 14:17:31
纺织科学研究(2017年7期)2017-07-25 07:48:49
渭南师范学院学报(2016年16期)2016-08-13 07:30:32
当代经济(2016年26期)2016-06-15 20:27:14
中国非金属矿工业导刊(2014年3期)2014-02-28 09:20:43
河南科技(2014年12期)2014-02-27 14:10:34
少儿科学周刊·少年版(2013年8期)2013-04-29 00:44:03
