
多聚腺苷二磷酸核糖聚合酶1调控β-1,4-甘露糖基转移酶介导甲状腺癌细胞增殖机制
2023-07-20 02:59刘梅黄晶张燕妮李珊胡绍波刘芳胡玉海孙文早
临床内科杂志 2023年6期
刘梅 黄晶 张燕妮 李珊 胡绍波 刘芳 胡玉海 孙文早
甲状腺癌(TC)的发病率逐年上升,已成为最常见的内分泌相关恶性肿瘤[1]。大多数甲状腺未分化癌(ATC)患者首次就诊时即发生远处转移,经传统治疗并不能明显提高ATC患者的生存率[2]。目前TC的发病机制尚未阐明[3]。但近年随着对TC分子生物学发病机制的深入研究,发现多种基因与TC的发病密切相关[4],这些基因的多靶点多激酶抑制剂已被开发并应用于TC的临床治疗,但部分患者疗效仍不理想[5]。因此,进一步发掘新型介导TC发病及进展的关键分子将为其治疗提供新方向。β-1,4-甘露糖基转移酶(ALG1)主要参与蛋白质糖基化修饰的初始阶段,前期在肝癌的研究中显示高表达的ALG1具有抑制肝癌细胞迁移的作用[6]。然而,ALG1在TC中的具体研究尚少。多聚腺苷二磷酸核糖聚合酶(PARP)1在DNA损伤修复过程中发挥关键作用[7]。研究表明,PARP1(Val762Ala)因缺乏多聚腺苷二磷酸(ADP)RNA酶活性而无法进行碱基切除修复,从而增加TC患病风险[8],但PARP1对TC发生发展的具体作用机制尚不明确。前期我们的研究表明,PARP1表达活化可能在TC的发展过程中发挥重要作用[9],本研究拟在此基础上,进一步探讨PARP1对TC细胞增殖、凋亡的影响,筛选出介导PARP1促进TC进展的关键糖基转移酶,从而为TC的靶向治疗提供理论依据。
材料与方法
1.材料:人正常甲状腺滤泡上皮细胞系(Nthy-ori 3-1,上海舜冉);TC细胞系(SW579及8305C)、人胚胎肾细胞(HEK293T,中国科学院细胞库);慢病毒表达载体(pLV)-EGFP/ALG1(武汉擎科);细胞培养基(RPMI1640)、细胞培养基(DMEM)、胎牛血清(美国Gibco);Trizol(美国Invitrogen);逆转录试剂盒、荧光定量PCR试剂盒(日本Takara);蛋白酶抑制剂、细胞计数试剂盒8(CCK-8)和二喹啉甲酸(BCA)蛋白浓度测定试剂盒(上海碧云天);PARP1、磷酸化蛋白激酶R样内质网激酶(pERK)、磷酸化丝裂原活化蛋白激酶p38(pp38)、磷酸化氨基未端蛋白激酶(pJNK)、磷脂酰肌醇3-激酶[PI3K(p110α)]、大鼠样肉瘤病毒癌基因(Kras)、ALG1和甘油醛-3-磷酸脱氢酶(GAPDH)多克隆抗体(武汉Proteintech);NMS-P118(美国Sigma-Aldrich)。全自动酶标仪(英国Biochrom);微量离心机(德国Eppendorf);流式细胞仪(美国BD);荧光显微镜(日本Olympus);凝胶成像系统(美国UVP)。
2.方法
(1)细胞培养:Nthy-ori3-1细胞系培养采用含10%胎牛血清和1%双抗(青霉素/链霉素)的RPMI1640培养基,TC细胞系培养采用含10%胎牛血清和1%双抗(青霉素/链霉素)的DMEM培养基,传代消化采用0.25%胰蛋白酶进行,培养环境为37 ℃、5%CO2。
(2)基因表达数据:下载肿瘤基因组图谱(TCGA) Thyroid Cancer(THCA)数据库的数据,采用BRB-Array Tools进行数据分析。共包含502例标本,其中甲状腺乳头状癌490例,其他病理类型12例。使用Kaplan-Meier数据库(http://kmplot.com)分析PARP1表达与总生存(OS)率之间的关系。采用R2:基因组学分析与可视化平台(http://r2.amc.nl)对TCGA TC数据集中与PARP1表达相关的前1 000个差异基因进行筛选分析,并对差异基因控制的前19个生物学过程的通路富集分析。
(3)蛋白质免疫印迹法(Western blot)检测蛋白表达水平:冰上裂解细胞样本,采用BCA法测定蛋白浓度。采用SDS-PAGE法经电泳、转膜后,与相应的一抗、二抗进行孵育,加入增强型化学发光试剂(ECL)发光剂,经UVP凝胶成像仪成像显影。在检测AGL1糖基化修饰实验中,转膜后用BSA封闭,生物素标记的MBL孵育后,加入辣根过氧化物酶(HRP)标记的链霉亲和素进行显影。
(4)CCK-8法检测细胞增殖情况:将对数期8305C细胞以1×103个/孔的密度接种到96孔板,相应处理后,加入10 μl/孔CCK-8后,避光再孵育4 h,用酶标仪检测450 nm处的吸光度值,其与活细胞数量成正比。
(5)8305C细胞过表达的构建:将人ALG1的cDNA克隆到pLV-EGFP过表达慢病毒载体构建pLV-ALG1过表达载体,引物序列:ALG1上游引物为5’-ATACCGGTAGCCAAGATGGCGGCCTCATG-3’,下游引物为5’-GCGTCGACTTCATCTCAAAACAAAATTTA-3’。阳性克隆经测序验证正确后,进行高纯度目的基因质粒抽提。将构建好的目的基因质粒和慢病毒共转染293T细胞,获得高质量浓缩慢病毒。取对数生长期的8305C细胞,按2×105个/孔接种于6孔板培养。对照组(pLV-EGFP组)与实验组(pLV-ALG1组)各加入4 μl浓缩慢病毒(滴度5×108TU/ml),经嘌呤霉素抗性筛选2天后扩增。荧光显微镜下观察感染后细胞状态及感染率,Western blot验证ALG1过表达效果。
(6)流式细胞术检测细胞凋亡情况:使用Accuri C6流式细胞仪进行检测。收集分别经DMSO和NMS-P118(1 μM)处理24 h的8305C细胞2×105个,加入195 μl Annexin V-FITC结合液重悬细胞,随后依次加入5 μl Annexin V-FITC和10 μl PI染液轻轻混匀,室温避光孵育15 min,随后冰浴、上机检测。
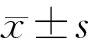
结 果
1.不同甲状腺细胞系中PARP1表达水平比较:两种TC细胞系(SW579和8305C)中PARP1表达水平高于正常细胞系Nthy-ori 3-1(1.43±0.28比0.42±0.15,2.17±0.33比0.42±0.15,P均<0.05)。
2.NMS-P118对TC细胞增殖凋亡的比较:NMS-P118+8305C细胞24 h、48 h及72 h后细胞增殖均低于同期的DMSO+8305C细胞(0.25±0.04比0.35±0.10,0.51±0.13比0.76±0.25,1.03±0.26比1.56±0.52,P均<0.05)。NMS-P118+8305C细胞24 h后细胞凋亡明显高于同期DMSO+8305C细胞(34.6±3.51比10.4±0.62,P<0.05)。
3.PARP1调控TC细胞生物信息学结果分析:PARP1高表达TC患者总生存期(OS)低于PARP1低表达TC患者(HR=3.21,95%CI1.11~9.27,P<0.05)。TCGA数据库中筛选出与PARP1表达相关的前1 000个差异基因(图1),并富集分析所调控的关键信号通路,结果显示除已被报道与PARP1功能相关的MAPK和Kras通路外,N-糖基化修饰信号通路中的基因显著富集(图2)。ALG1高表达患者OS具有降低倾向(HR=4.24,95%CI1.25~14.27,P<0.05)。

图1 TCGA数据库PARP1表达显著相关的前1 000个差异基因
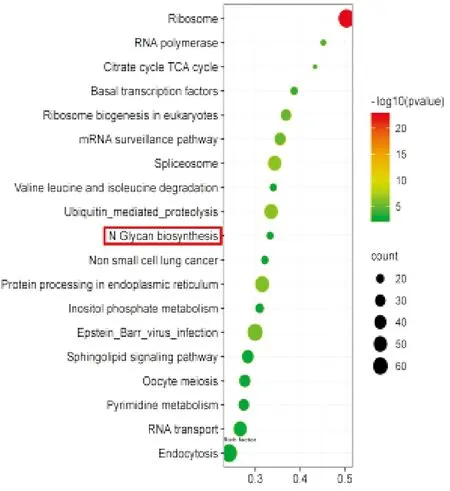
图2 差异基因的富集分析
4.PARP1调控TC细胞机制的验证:NMS-P118显著抑制ERK、JNK和Kras的活化水平(0.61±0.12比0.08±0.03,1.24±0.15比0.31±0.08,3.07±0.25比0.86±0.18,P均<0.05),但对p38的影响不明显(0.35±0.10比0.29±0.08,P>0.05)。NMS-P118显著抑制ALG1表达(1.21±0.25比0.28±0.07),显著下调8305C细胞的甘露糖修饰水平(0.57±0.21比0.26±0.06,P均<0.05)。
5.不同表达水平ALG1对TC细胞增殖凋亡的比较:过表达ALG1可明显逆转NMS-P118+8305C细胞24 h、48 h及72 h的细胞增殖作用(0.41±0.02比0.24±0.08,0.84±0.05比0.52±0.13,1.60±0.18比1.03±0.24,P均<0.05)。过表达ALG1k可逆转NMS-P118促进TC细胞凋亡的作用(5.46±0.48比35.99±4.48,P<0.05)。
讨 论
抑制凋亡在TC的进展中具有关键作用,如磷脂酶Cδ3(PLCD3)通过Hippo通路促进TC细胞的增殖、迁移和侵袭,从而抑制其凋亡,进而促进TC进展[10]。虽然PARP1在多种肿瘤中具有促凋亡作用[11-12],但研究显示PARP1对卵巢癌等肿瘤的发展具有促进作用[13]。为探究PARP1在TC中的作用,我们通过对正常甲状腺细胞系和两种TC细胞系进行PARP1检测,发现PARP1在两种TC细胞系中均高水平表达。同时TCGA数据库分析也显示PARP1高表达TC患者OS低于PARP1低表达的TC患者,这说明TC中PARP1高水平表达预示不良预后,可能有利于TC进展。此外,我们使用NMS-P118处理8305C细胞后,检测细胞增殖与凋亡状况,结果显示抑制PARP1的表达可显著抑制TC细胞的增殖并促进其凋亡,这进一步表明PARP1可能在促进TC细胞增殖而加快TC进展过程中发挥着重要作用。
在研究PARP1影响TC细胞生物学功能具体机制的过程中,我们通过从TCGA数据库中筛选出与PARP1表达呈显著相关的前1 000个差异基因,并通过富集分析找出其所调控的主要信号通路。结果显示,PARP1参与调节TC细胞中包括剪接体、泛素化蛋白水解、RNA转运和内吞作用在内的多种生物学过程。PARP1调节TC细胞生物学过程的多面性既为我们靶向PARP1治疗TC提供了更多的可能性,又让我们的研究方向充满了复杂性。本研究中我们发现PARP1下游调控的ERK、JNK信号通路及N-糖基化修饰都可能在TC细胞中发挥抑制凋亡的作用。且我们使用PARP1抑制剂也证实抑制PARP1表达可显著抑制TC细胞中ERK和JNK的磷酸化水平。研究表明PARP1不但通过MAPK促进卵巢癌的进展[14],还在非小细胞肺癌通过MAPK通路促进其转移[15]。值得注意的是,虽然最近的研究发现p38介导了核蛋白Kin17对TC细胞增殖和迁移的促进作用[16],但本研究中PARP1抑制剂对TC细胞p38的激活无明显抑制作用,因此PARP1可能通过独立于p38的MAPK通路在TC细胞中发挥促癌作用,具体机制有待进一步深入探究。
除了常规信号通路,本研究还重点关注了N-糖基化修饰途径。N-糖基化修饰是真核生物中一种高度保守的蛋白质修饰过程,多种甘露糖基转移酶参与N-糖基化途径并在不同的肿瘤当中发挥促癌作用[17]。但是,不同的唾液酸转移酶对肿瘤的进展具有不同作用,如ST3GAL1能够促进前列腺癌细胞增殖、侵袭,抑制其凋亡[18],而ST6GAL1则通过增加细胞间黏附因子1(ICAM-1)的唾液酸化而增加其蛋白质的稳定性,抑制结直肠癌细胞的转移能力[19]。我们通过TCGA数据库对TC患者中ALG1的表达与预后情况进行分析,发现ALG1高表达的TC患者OS具有降低倾向。此外,过表达ALG1可逆转NMS-P118对TC细胞的抑制作用。这说明N-糖基化修饰可能在TC的发展过程中扮演着重要角色,且有可能受PARP1的影响。但ALG1通过N-糖基化修饰下游何种关键靶分子进而影响TC细胞功能,还有待进一步的实验探索。
综上所述,本研究表明TC中高度表达的PARP1通过调节N-糖基化酶ALG1促进TC细胞的N-糖基化修饰,从而抑制TC细胞凋亡,促进其增殖。高水平表达的PARP1和ALG1都预示TC患者OS降低,这将为靶向PARP1治疗TC提供重要理论依据。
猜你喜欢
现代畜牧科技(2021年6期)2021-07-16
心电与循环(2020年1期)2020-02-27
江苏农业科学(2017年5期)2017-04-15
山东医药(2015年14期)2016-01-12
医学研究杂志(2015年12期)2015-06-10
江苏大学学报(医学版)(2015年2期)2015-04-17
中国医药导报(2015年26期)2015-02-28
湖北农业科学(2014年3期)2014-07-21
中国粮油学报(2014年7期)2014-02-06
现代检验医学杂志(2014年1期)2014-02-06
