
慢性肾脏病与高尿酸血症
2023-07-20 03:18周涵梁伟
临床内科杂志 2023年6期
周涵 梁伟
慢性肾脏病(CKD)是由于各种原因导致的肾脏结构与功能损伤,可引发多种并发症(包括矿物质代谢改变、贫血、代谢性酸中毒和心血管事件)[1]。CKD患者发生透析、心血管事件和死亡风险的升高,全球CKD患病率约为13.1%,2023年调查结果显示我国CKD患病率为8.2%[2-4]。目前,CKD缺乏有效的治疗方法,临床医生只能针对危险因素和并发症进行治疗。早期识别CKD的危险因素有助于延缓疾病进展,改善患者生活质量和预后。随着经济的快速发展和饮食方式的改变,我国高尿酸血症(HUA)患病率也在不断上升。HUA是CKD常见的临床表现,近年来,多项观察性研究结果显示,HUA不仅是CKD患者肾功能下降的标志物,也是CKD进展的独立危险因素[3,5]。HUA与CKD、糖尿病肾脏疾病(DKD)、微小病变肾病、IgA肾病等多种肾脏疾病的发生与发展密切相关[6-7]。尿酸可通过炎症、氧化应激、激活肾素血管紧张素(RAS)系统、促进肾脏纤维化等机制损伤肾脏[8]。在CKD患者中,HUA与CKD的先后关系并不是十分明确,确定两者之间因果关系存在一定难度。尿酸作为一种可改变的代谢产物,可能是延缓疾病进展的治疗靶点,深入认识无症状HUA对于肾脏的损害,进行适当的干预是有意义的。本文主要阐述HUA与CKD之间的关系、尿酸在CKD进展中的作用及降尿酸治疗在延缓疾病方面的作用。
一、HUA的患病率
随着经济的快速发展及饮食方式的改变,普通人群中HUA的患病率在不断上升。不同地区HUA的患病率见表1。一项全国性抽样调查研究结果显示,2015~2016年美国男性HUA患病率为20.2%,女性为20.0%[9]。发达国家HUA患病率通常高于经济发展中国家。相关研究显示,我国HUA的患病率也在快速上升,且呈年轻化趋势,2015~2016年我国的HUA总患病率为11.1%(男性19.3%、女性2.8%),2018~2019年为14.0%(男性24.4%、女性3.6%)[10]。我国幅员辽阔,不同地区的HUA患病率也存在差异。一项前瞻性队列研究结果发现,2017~2018年京津冀地区HUA的总体患病率为19.37%[11],高于全国总体患病率,这可能与经济发展程度相关;河南农村人群HUA的患病率为10.24%[12]。HUA的患病率在性别上也存在差异,绝经期前的女性患病率低于男性,这与雌激素增加尿酸排泄有关。相关研究结果表明雌激素可抑制肾脏中的尿酸盐重吸收转运蛋白(URAT1)活性,减少尿酸重吸收水平[13]。
表1 不同地区HUA的患病率
肾脏是尿酸排泄的主要器官,血尿酸会随着eGFR下降而升高,CKD患者HUA的患病率更高。终末期肾病(ERSD)患者HUA的患病率超过了60%。一项流行病学调查结果显示,在CKD3~5期患者中,HUA的患病率达到了70%~85%[27]。有研究结果发现,即使在肾功能正常的情况下,IgA肾病患者HUA的发生率也较高,约为40.9%[28]。DKD患者HUA的发生率为32.4%,不同肾脏疾病患者中,HUA的患病率存在差异,特别是在IgA肾病中较高,相关机制尚不清楚。
二、尿酸在肾脏中的代谢
人体中每日产生的尿酸有70%~80%经过肾脏排泄,肾脏排泄尿酸的过程分为4步:(1)肾小球的滤过;(2)肾小管的重吸收;(3)肾小管的再分泌;(4)分泌后的重吸收。
在肾功能正常的情况下,尿酸盐在肾小球几乎100%滤过,尿酸的重吸收和分泌主要在近端肾小管。重吸收、分泌、分泌后再吸收分别在近端肾小管S1段、S2段及S3段进行[29]。尿酸在肾脏的代谢受尿酸转运相关蛋白的调节[30],尿酸的重吸收是由位于肾小管上皮细胞顶端的URAT1和位于顶端和基底侧的葡萄糖转运蛋白9(GLUT9)完成,分泌由位于基底侧的有机阴离子转运蛋白(OAT)1和OAT3、位于顶端的ATP结合转运体G2(ABCG2)和钠依赖性无机磷酸盐转运蛋白(NPT)1和NPT4完成。URAT1是HUA发病的关键机制之一,也是一些药物治疗HUA的作用靶点[31]。有研究结果表明,GLUT9在尿酸重吸收过程中起着重要作用,甚至可能比URAT1的作用更为明显[32]。肾脏对血尿酸的稳态起着至关重要的作用,了解这些转运蛋白的调节机制可为未来高尿酸血症提供更多的治疗靶点。
三、HUA与CKD的关系
在肾功能正常人群中,HUA可预测CKD的发生;而在CKD患者中,HUA可预测CKD的进展。一项前瞻性研究共纳入34 831例CKD患者,基线时13.4%的患者有HUA,中位随访时间为4.1年,该研究表明血尿酸水平升高是CKD发生的1个危险因素,尿酸每增加1 mg/dl,CKD的发生风险增加15%(HR=1.15,95%CI1.08~1.24,P<0.001)[3]。一项人群随访研究也得出了类似的结论,且HUA与女性CKD的发生相关性更高[5]。Srivastava等[33]研究了3 885例CKD2~4期的患者,中位随访时间为7.9年,发现在eGFR≥45 ml·min-1·(1.73 m2)-1的患者中,血尿酸水平升高增加了ESRD的风险,提示HUA是CKD进展的独立危险因素。
有研究评估了在亚组中HUA与肾功能下降之间的相关性,包括DKD、IgA肾病和微小病变肾病等。Pilemann-Lyberg等[34]研究了1型糖尿病患者血尿酸与eGFR下降之间的相关性,中位随访时间为5.3年,发现基线时血尿酸水平升高与eGFR下降相关(HR=3.18,95%CI1.71~5.93,P<0.001)。IgA肾病患者HUA是预后不良的1个独立危险因素,且与IgA肾病肾小管损伤密切相关。一项回顾性研究分析了4 339例IgA肾病患者,中位随访时间为6.1年,cox比例风险模型提示HUA是IgA肾病进展的独立危险因素,且在女性患者中的作用更大[35]。微小病变肾病患者HUA是进展为ESRD的独立危险因素,基线时患者的血尿酸水平每增加29 mg/dl,微小病变肾病进展的风险增加1%[36]。HUA是肾功能下降常见的临床表现,血尿酸随着eGFR下降而升高,而HUA可通过多种机制加重肾损伤,形成恶性循环。在多种危险因素和CKD主要发病机制(如蛋白尿和高血压)的背景下,血尿酸的不利作用难以确定。越来越多的基础和临床观察性研究证实了HUA在CKD发生、进展中的作用。部分研究结论不一致可能与研究纳入了CKD 4~5期患者有关,CKD4~5期的患者中,肾损伤严重,疾病进展迅速,相比之下血尿酸的不利作用并不显著。
四、尿酸在CKD进展中的作用机制
1.促进炎症反应:人体血尿酸水平>420 μmol/L时,可在组织中沉积,引发炎症反应。单尿酸盐晶体可激活NOD样受体蛋白3(NLRP3)炎症小体,进而促进炎症细胞分泌活性IL-1β和IL-18等炎症因子[37]。既往认为单尿酸盐晶体是导致HUA肾损伤的主要原因,最近有研究结果表明可溶性尿酸盐可通过多种机制导致肾损伤,可溶性尿酸盐也可激活NLRP3炎症小体,促进炎症因子的分泌,导致炎症反应发生[38-39]。可溶性尿酸盐可激活肾小管上皮细胞中的核因子(NF)-κB信号传导,促进肿瘤坏死因子(TNF)-α、TNF-β1、单核细胞趋化蛋白1(MCP-1)等炎症因子表达[40]。在肾小管细胞上,可溶性尿酸盐与其表面受体Toll样受体4(TLR4)结合,诱导下游MCP-1的表达升高[41],刺激炎症反应,诱发肾小管损伤和肾间质纤维化。
2.氧化应激反应:可溶性尿酸盐在细胞外可作为抗氧化剂清除自由基和活性氧(ROS),而进入细胞内可能是一种促氧化剂,导致肾损伤的主要因子为细胞内尿酸盐。激活还原型烟酰胺腺嘌呤二核苷酸磷酸氧化酶(NOX)是可溶性尿酸盐促进氧化应激反应的关键机制。黄嘌呤氧化还原酶氧化生成尿酸时的副产物之一是ROS,过量的ROS可导致氧化应激反应,并激活NF-κB信号途径促进细胞炎症反应的发生。肾小管上皮细胞中线粒体余量丰富,可溶性尿酸盐引起的氧化应激反应可导致线粒体的功能障碍,增加下游凋亡蛋白(如p53、Bax、Caspase-9/-3等)的表达,导致肾小管上皮细胞凋亡[42]。Yang等[43]通过建立尿酸性肾病大鼠模型发现,HUA组中24小时尿蛋白、血清肌酐(SCr)和血尿酸水平与对照组比较显著增加。且HUA组的氧化应激指标增加、线粒体中凋亡蛋白显著表达、肾小管上皮细胞大量凋亡。
3.RAS系统激活:RAS系统激活是尿酸引起肾血管病变的主要机制之一。基础研究结果表明,在体外可溶性尿酸盐激活RAS系统,导致肾素、血管紧张素Ⅱ等表达上调,从而促进内皮细胞的损伤、衰老和凋亡[44]。有研究表明,尿酸盐可能通过激活有丝分裂原、活化蛋白激酶的途径来激活RAS系统,从而减少一氧化氮(NO)合成,诱导血管内皮功能障碍,导致肾小球高血压,减少灌注,最终导致肾小球硬化[45]。观察性研究证实,在IgA肾病患者中,HUA与其肾小球硬化程度密切相关[6,46]。在大鼠模型中,果糖来源的尿酸被证明可激活肾内RAS系统,导致肾小管Na+转运蛋白表达增加,引起盐敏感性高血压[47]。RAS系统激活后,血管紧张素Ⅱ增加,与近端肾小管细胞表达的TLR4相互作用[48],促进肾脏的炎症反应和氧化应激反应。炎症反应是CKD进展的驱动因素之一,炎症与氧化应激相互作用,而RAS系统可促进炎症反应及氧化应激反应。
4.促进纤维化:肾小管间质纤维化是各种肾脏疾病的共有病理表现。有研究发现HUA是IgA肾病节段性肾小球硬化和肾小管萎缩/间质纤维化的独立危险因素[28]。肾小管上皮间充质转化(EMT)是肾纤维化的关键[49],上皮细胞转化为间充质细胞,分化为成纤维细胞和肌成纤维细胞,肌成纤维细胞产生基质,沉积的细胞外基质可导致肾小管间质纤维化。除氧化应激和炎症反应外,尿酸盐也可直接影响肾小管上皮细胞表型转化。可溶性尿酸盐可激活TLR4/NF-κB信号通路,促进肾小管上皮细胞的EMT[50]。体外实验结果证实,可溶性尿酸盐可诱导氧化应激和糖萼脱落,从而促进内皮细胞间质转化(Endo-MT)[51]。可溶性尿酸盐可增加转化生长因子-β1(TGF-β1)表达和Smad3磷酸化的上调,通过TGF-β1/Smad3信号通路来增加基质蛋白合成并减少其降解,促进肾小管间质纤维化的发生和发展[52]。肾小管间质纤维化发生的一个关键机制就是TGF-β1/Smad3信号通路的激活。
五、降尿酸治疗对CKD的影响
由前文可知,大部分基础及观察性研究结果证明尿酸是CKD发生和进展的1个独立危险因素,降尿酸治疗应是一种可延缓疾病进展的方法。一些小样本的随机对照研究(RCT)和观察性研究报告了降尿酸治疗在预防或延缓CKD患者肾功能下降方面的作用[53-54]。一项单中心、单盲、RCT评估了113例eGFR<60 ml·min-1·(1.73 m2)-1的患者,中位随访时间为84个月,别嘌呤醇组肾脏进展事件发生率低于对照组(HR=0.32,95%CI0.15~0.69,P=0.004)。但最近部分RCT发现,降尿酸治疗后并未改善患者肾脏预后[55]。一项RCT评估了别嘌呤醇在1型糖尿病和eGFR为40~99.9 ml·min-1·(1.73 m2)-1的糖尿病肾脏疾病中的作用,与安慰剂相比,别嘌呤醇在延缓eGFR下降方面差异无统计学意义[56]。另一项RCT研究纳入了269 651例肾功能正常的CKD患者,其中10.9%接受了降尿酸治疗,但没有改善肾脏预后[GFR<60 ml·min-1·(1.73 m2)-1、白蛋白尿、ERSD],降尿酸治疗没有发挥预防作用[57]。同样,在CKD 3~4期的患者中,降尿酸治疗没有发挥延缓CKD进展的作用[56]。降尿酸治疗对CKD影响的RCT研究见表2,研究中的降尿酸治疗药物大部分是黄嘌呤氧化酶抑制剂(别嘌呤醇、非布索坦),其为HUA治疗的一线药物。有观察性研究结果表明,非布索坦在延缓肾功能下降方面优于别嘌呤醇[58]。但还需要更多大型RCT探讨非布索坦及排尿酸药物在延缓肾脏疾病方面的作用。临床上对CKD伴HUA的患者是否可进行降尿酸治疗尚无统一的结论。基础及观察性研究结果表明,理论上降尿酸治疗应可延缓患者的肾脏进展,但目前的研究结果提示并非所有CKD患者都能够从中获益。部分研究结论不一致有几种解释:(1)一些RCT的样本量小、随访时间短,降尿酸治疗在短期内的获益可能不明显;(2)不同研究关于HUA、CKD进展的定义不一致,存在异质性;(3)CKD是由多种肾脏疾病导致的慢性疾病,治疗方案存在差异,影响CKD进展的因素较多,降尿酸治疗的效果难以显现。
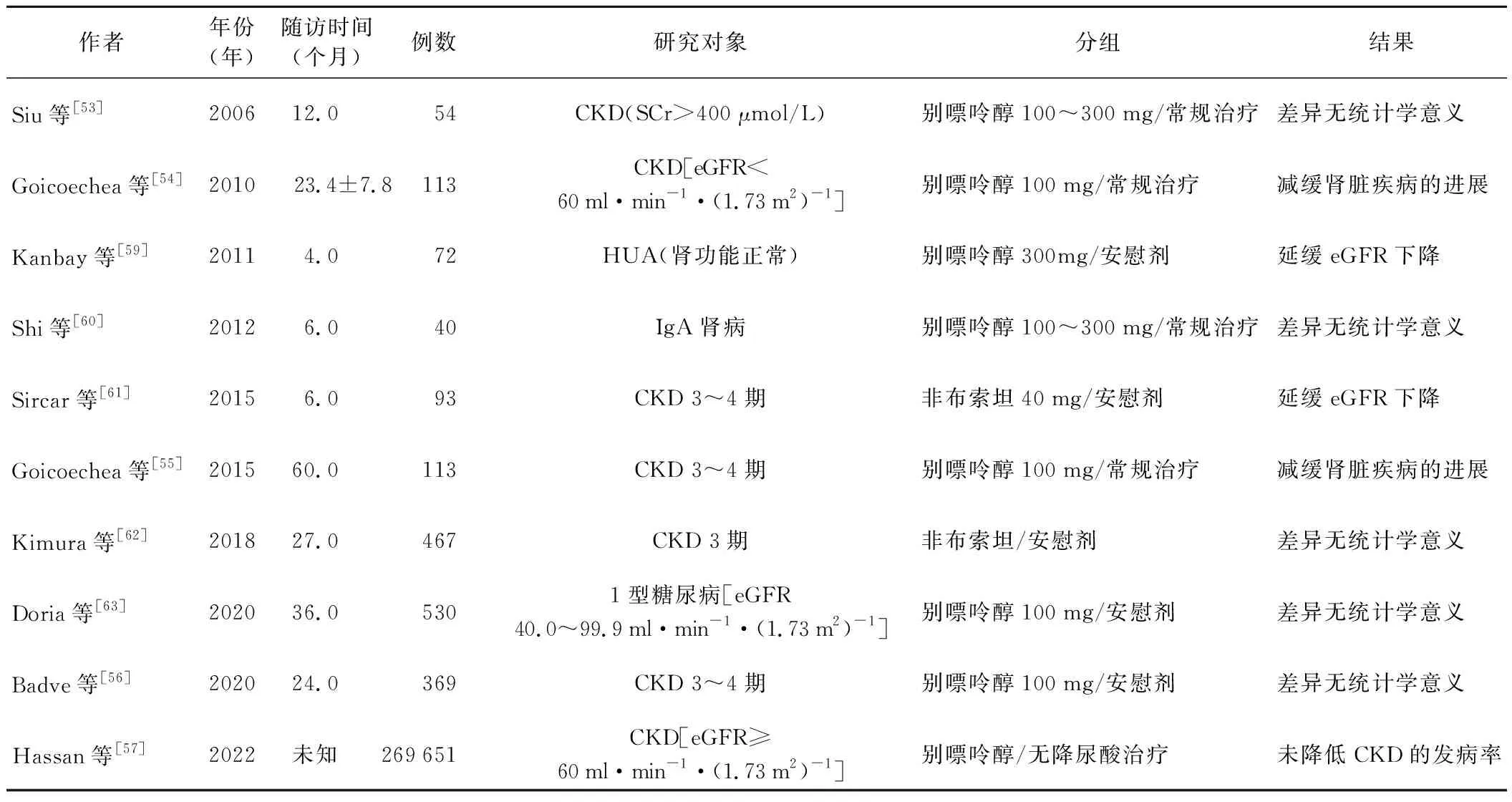
表2 降尿酸治疗对CKD影响的RCT研究
六、总结
CKD的发病率较高,并发症较多,总体预后不良。HUA对CKD的影响一直存在争议,但越来越多的研究证实高尿酸血症是CKD发生和发展的1个独立危险因素,且与多种肾脏疾病进展相关,高尿酸血症可通过炎症反应、氧化应激反应、RAS系统激活、促进纤维化等机制损伤肾脏。目前,降尿酸治疗获益仍存在争议,未来需要更多大型RCT来确定干预时机和阈值。
猜你喜欢
中老年保健(2022年4期)2022-08-22
健康体检与管理(2022年2期)2022-04-15
现代临床医学(2021年2期)2021-03-29
昆明医科大学学报(2021年1期)2021-02-07
益寿宝典(2018年5期)2018-01-28
中国男科学杂志(2016年9期)2016-03-20
中西医结合心脑血管病杂志(2016年20期)2016-03-01
中外医疗(2015年11期)2016-01-04
中国健康心理学杂志(2015年5期)2015-09-05
医学研究杂志(2015年8期)2015-06-22
