
伪狂犬病病毒gE/gI/TK基因缺失对感染PK-15细胞的差异表达蛋白质组分析
2023-07-08 12:34张洪亮王凤雪刁志凯李桂梅温永俊
中国兽医学报 2023年5期
张洪亮,王凤雪,刁志凯,李桂梅,黄 娟,温永俊*,单 虎*
(1.青岛农业大学 动物医学院 山东省预防兽医学重点实验室,山东 青岛 266109; 2.内蒙古农业大学 兽医学院 农业部动物疾病临床诊疗技术重点实验室,内蒙古 呼和浩特 010018)
伪狂犬病病毒(pseudorabies virus,PRV)是疱疹病毒科(Herpesviridae)水痘疱疹病毒属(Varicellovirus)的成员,又称为猪疱疹病毒I型(suid herpesvirus 1,SuHV1)[1]。PRV是引起伪狂犬病(pseudorabies,PR),即奥耶斯基病(Aujeszky's disease,AD)的病原体,可导致幼仔猪的神经系统和呼吸系统疾病,以及胎儿死亡和/或妊娠母猪流产,并导致种公猪的繁殖能力降低[2]。PRV可跨多物种传播,除了感染猪(唯一天然宿主),还可感染反刍动物(如牛羊)、食肉动物(如水貂、狐狸)和啮齿动物(如小鼠)等各种野生和家养动物,并有报道发现PRV可感染人,导致人眼内炎和脑炎,危害公共卫生安全[3-4]。
自从2011年PRV变异株出现后,导致我国多个省份和地区已免疫Bartha-K61株疫苗的猪场发生PR疫情[5-6]。有研究表明,Bartha-K61株疫苗对变异株的保护力有所降低,尽管该观点还存在争议[5,7]。PRV具有大约150 kb的双链DNA基因组,病毒粒子结构复杂。PRV不同毒株在宿主致病性和诱导免疫反应中存在显著差异,目前尚不完全了解PRV感染的致病机制,以及PRV与宿主细胞之间的相互作用[8]。为了解PRV变异株的主要毒力基因gE/gI/TK缺失后对感染宿主细胞的影响,利用串联质谱标签(tandem mass tags,TMT)标记技术,结合高精度质谱仪串联分析,分别对PRV SD-2017株、SD-2017ΔgE/gI/TK株和Bartha-K/61株感染的PK-15细胞进行了差异表达蛋白质组的生物信息学分析,以期为PRV致病和感染机制研究提供新的思路或方向。
1 材料与方法
1.1 材料
1.1.1病毒与细胞 PRV SD-2017变异株、PRV Bartha-K/61疫苗株、PK-15细胞系,均由本实验室保存。PRV SD-2017ΔgE/gI/TK株系本实验室利用SD-2017变异株构建的gE、gI和TK 3基因缺失株。
1.1.2主要试剂 胎牛血清(FBS)、DMEM培养基、胰酶购自Gibco公司;Bradford蛋白定量试剂盒购自Bio-Rad公司;TMT质量标记试剂盒购自Thermo公司;考马斯亮兰R-250购自Amresco公司;PVDF膜购自Millipore公司;MYLPF、ERI1、XRCC6和GAMT抗体购自Proteintech公司;β-tublin抗体购自万类公司;TanonTMHigh-sig ECL Western Blotting Substrate购自Tanon公司。
1.1.3主要仪器 Axio Vert.A1倒置显微镜(蔡司公司,德国);Scients-04生物安全柜(苏州安泰空气技术有限公司);CCL-170B-8 CO2培养箱(ESCO公司,新加坡);EASY-nLCTM1200纳升级UHPLC系统、L-3000 HPLC系统、Q ExactiveTMHF质谱仪(赛默飞世尔公司,美国);Vilber Fusion FX6多功能成像系统(Vilber公司,法国)。
1.2 方法
1.2.1细胞培养及病毒接种 PK-15细胞用改良的Eagle培养基(含10% FBS的DMEM)培养于37℃、 5% CO2、饱和湿度的培养箱中。将PK-15单层细胞用0.25%胰蛋白酶和0.02%乙二胺四乙酸(EDTA)分散并接种在6 cm细胞培养瓶中。待细胞密度达到70%时,用PBS洗涤细胞2次,分别接种MOI=0.1的PRV各毒株,吸附1 h后加入含2% FBS的DMEM继续培养。未感染的PK-15细胞用作对照组。在感染后24 h收集PRV或对照组的细胞,每组设3个独立的生物学重复。
1.2.2蛋白质提取及质量浓度检测 感染PRV和未感染的PK-15细胞弃去DMEM培养液,用冷的PBS洗涤3次。用胰酶消化使贴壁细胞脱落,1 000×g离心10 min收集悬浮细胞。样品中加入600 μL lysis buffer (100 mmol/L 碳酸氢铵、6 mol/L 尿素、0.2% SDS,pH=8),超声波破碎5 min,然后4℃、12 000×g离心15 min,加入4倍体积的冷丙酮(含有10 mmol/L DTT)沉淀2 h。4℃、12 000×g离心15 min后,收集沉淀。加入800 μL冷丙酮(含有10 mmol/L DTT)重悬沉淀,破坏蛋白质的二硫键。4℃、12 000×g离心15 min后收集干燥的沉淀物,并用600 μL 8 mol/L Urea(50 mmol/L Tris buffer,8 mol/L Urea,pH=8)溶解。按照Bradford蛋白定量试剂盒说明书测定蛋白的质量浓度。
1.2.3TMT试剂标记及质谱分析 各组取120 μg蛋白样品,按照Thermo公司TMT质量标记试剂盒说明书进行标记。使用L-3000 HPLC系统(RIGOL)进行馏分分离,色谱柱为Waters BEH C18 (4.6 mm×250 mm,5 μm)。使用EASY-nLCTM1200纳升级UHPLC系统进行液质检测,预柱为自制预柱(2 cm×75 μm,3 μm),分析柱为自制分析柱(15 cm×150 μm,1.9 μm)。使用Q ExactiveTMHF质谱仪进行质谱分析,扫描范围为350~1 500 m/z,生成质谱检测原始数据(.raw)。
1.2.4数据分析 将原始的MS/MS数据直接导入到Proteome Discoverer 2.2软件(PD 2.2,Thermo)进行数据库检索,分析谱肽和蛋白定量。使用蛋白质数据库Sus_scrofa_uniprot_2020_1_8.fasta (120594 sequences)搜索MS/MS数据。对于标准化后使用TMT测得的蛋白质丰度比率,我们专门使用P<0.05的比率,只有差异倍数FC变化>1.2或<0.83被认为是显著的。
1.2.5生物信息学分析 使用基因本体功能注释(GO,http://geneontology.org/)对鉴定的和差异表达的蛋白质序列进行定位分析。使用直系同源群簇(COG,http://www.ncbi.nlm.nih.gov/COG/)和京都基因与基因组百科全书(KEGG,http://www.kegg.jp/kegg/pathway.html)数据库分析蛋白质家族和途径。
1.2.6蛋白质印迹分析验证 在感染后24 h收集PRV感染组细胞和对照组细胞。将来自每个样品的等量细胞裂解液与5×上样缓冲液混合,煮沸10 min,用12% SDS-PAGE分离蛋白并转移至PVDF膜。室温下PVDF膜在含5%脱脂牛奶的PBST中封闭2 h,然后在4℃下与一抗孵育过夜。将膜用TBST洗涤3次,并与辣根过氧化物酶(HRP)偶联的二抗在室温下孵育1 h。最后,通过添加ECL Western blot化学发光底物使蛋白质条带可视化,并使用Fusion FX6进行成像观察。
2 结果
2.1 蛋白质的质谱定量分析及鉴定结果本研究基于TMT标记结合LC-MS/MS对接种PRV SD-2017株、SD-2017ΔgE/gI/TK株和Bartha-K/61疫苗株感染后24 h的PK-15细胞内蛋白表达差异进行了检测分析。亲本毒株PRV SD-2017与SD-2017ΔgE/gI/TK比较,分析出6 013个蛋白,共鉴定到19个差异表达蛋白,其中12个蛋白上调,7个蛋白下调;SD-2017ΔgE/gI/TK(R_SD2017)与传统疫苗株Bartha-K/61比较,分析出4 690个蛋白,共鉴定到176个差异表达蛋白,其中157个蛋白上调,19个蛋白下调。各组主要差异表达蛋白质见表1,2。

表1 PRV SD-2017与SD-2017ΔgE/gI/TK感染组相比主要差异表达蛋白

表2 PRV SD-2017ΔgE/gI/TK与Bartha-K/61感染组相比主要差异表达蛋白
2.2 差异蛋白质的GO分析为了进一步扩展差异表达蛋白质的分子特征,搜索了Gene Ontology和UniProt数据库,并将这些蛋白质分配给了不同的生物学过程、分子功能和细胞成分。PRV SD-2017与SD-2017ΔgE/gI/TK比较(图1),差异蛋白主要参与肽交联、钙离子跨膜转运、糖酵解等生物学过程;差异蛋白主要分布在细胞质、细胞内、大分子复合物等部位;差异蛋白分子功能主要为核苷-三磷酸酶活性、金属离子结合、钙离子结合等。
PRV SD-2017ΔgE/gI/TK与Bartha-K/61比较(图2),差异蛋白主要参与核酸代谢、RNA代谢、RNA加工等生物学过程,差异蛋白主要分布在核膜、核质、细胞内膜性细胞器等部位,差异蛋白的分子功能主要为结合、杂环化合物结合、核酸结合等。
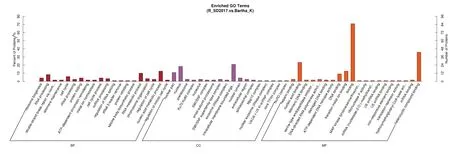
图2 PRV SD-2017ΔgE/gI/TK与Bartha-K/61感染PK-15细胞的差异蛋白质的GO分析
2.3 差异蛋白质的KEGG通路分析KEGG数据库用于识别参与差异表达蛋白质的代谢途径。与PRV SD-2017感染的PK-15 细胞相比,SD-2017ΔgE/gI/TK比较感染PK-15细胞的差异蛋白主要与紧密连接、糖酵解/糖异生等、RNA降解途径有关(图3)。但与Bartha-K/61感染的PK-15细胞相比,SD-2017ΔgE/gI/TK感染的PK-15细胞的差异蛋白主要与真核生物中的核糖体生物发生、RNA降解、黏着斑、B细胞受体信号通路等途径有关(图4)。

图3 PRV SD-2017与SD-2017ΔgE/gI/TK感染的PK-15细胞的差异蛋白KEGG通路分析
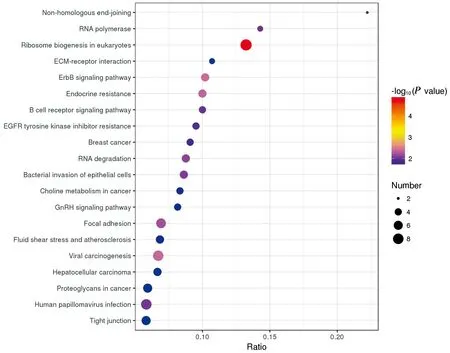
图4 PRV SD-2017ΔgE/gI/TK与Bartha-K/61感染的PK-15 细胞的差异蛋白KEGG通路分析
2.4 差异蛋白的Western blot验证为验证通过TMT分析鉴定出的差异表达蛋白,分别选择了基于比例的蛋白进行Western blot分析。感染PRV SD2017和SD-2017ΔgE/gI/TK细胞之间2种蛋白质(MYLPF和ERI1)的比率与从TMT方法获得的比率一致(图5A)。感染SD-2017ΔgE/gI/TK和Bartha-K/61细胞之间2种蛋白质(XRCC6和GAMT)的比率与从TMT方法获得的比率一致(图5B),表明本研究的蛋白组学定量结果准确可信。
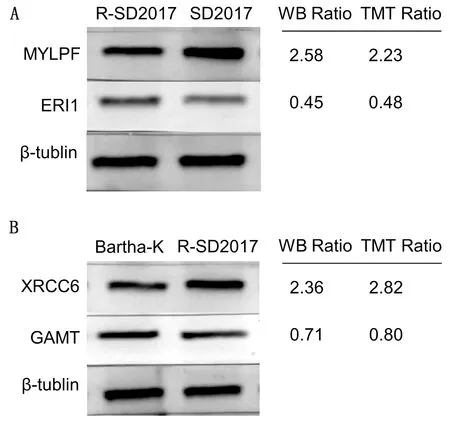
A.PRV SD2017和SD-2017ΔgE/gI/TK感染PK15 细胞中MYLPF、ERI1蛋白的Western blot和TMT比率(SD2017/R-SD2017);B.PRV SD-2017ΔgE/gI/TK和Bartha-K/61感染PK15细胞中XRCC6和GAMT蛋白的Western blot和TMT比率(R-SD2017/Bartha-K)。β-tublin蛋白用作标准化和上样对照
3 讨论
由于TMT标记定量蛋白质组学技术通量高、准确度高、鉴定效率高,可对多个样品进行差异表达蛋白的定量分析,为促进病毒-宿主相互作用的解析提供了有效的工具,已应用于多种动物病毒的蛋白质组学研究[9-10]。本研究通过生物信息学分析发现,PRV gE/gI/TK 3个毒力基因缺失后,与亲本毒株相比,SD-2017ΔgE/gI/TK株表现出ATP2A1、KDM8、Eri1和TGM2等多种蛋白质的差异表达。这些差异蛋白与细胞凋亡、免疫反应、疾病状态等密切相关。其中ATP2A1参与致癌和凋亡过程[11]。KDM8是一种H3K36me2组蛋白去甲基化酶,作用于细胞周期蛋白A1编码区以调节癌细胞增殖[12]。Eri1是一种进化上保守的3′-5′外切核糖核酸酶,参与5.8S rRNA 3′末端加工和复制依赖性组蛋白mRNA的转换,作为核糖体和组蛋白mRNA相关蛋白的成员被招募到不同的RNA代谢途径中[13]。而且Eri1调节淋巴细胞中的miRNA稳态,是正常NK细胞发育和抗病毒免疫所必需的[14]。转谷氨酰胺酶TGM2是一种多功能酶,具有转谷氨酰胺酶交联、G蛋白信号传导和激酶活性,据推测在癌症、神经退行性改变等许多疾病中发挥作用[15]。
与Bartha-K/61传统疫苗株相比,SD-2017ΔgE/gI/TK株的差异表达蛋白更加复杂。其中上调蛋白XRCC6不仅可以调节人类T淋巴细胞病毒1型(HTLV-1)的复制,还可以调节DNA病毒介导的先天免疫反应[16-17]。上调的核仁蛋白SURF6参与了核糖体生物发生过程中rRNA加工的各个步骤,是哺乳动物细胞增殖和G1/S转变的新型正调节因子,暗示SURF6是一种潜在的致癌蛋白[18]。另外,上调蛋白RRP1B是某些E2F1促凋亡靶基因的表达和DNA损伤剂诱导细胞凋亡所必需的[19]。而下调蛋白GrpEL1是一种核苷酸交换因子,可帮助mtHSP70在线粒体中进行非天然折叠的蛋白,促进神经元线粒体的稳态[20]。本研究还发现与自噬有关的DNM2和PYCR2蛋白下调。自噬体前体的释放是由DNM2依赖性断裂介导的,该过程受DNM2与LC3结合的调节,并因自噬诱导刺激而增加[21]。先前的研究表明,PYCR2的消耗与自噬的诱导有关,ASFV E199L蛋白通过与PYCR2相互作用诱导完全自噬并下调PYCR2的表达水平,为自噬在ASFV感染过程中的作用提供了有价值的参考[22]。而且,PYCR2缺失通过SHMT2增加大脑中的甘氨酸水平导致神经退行性变[23]。
综上,本研究基于TMT技术对感染PRV SD-2017ΔgE/gI/TK株PK-15细胞的差异蛋白质组进行了全面分析,将其与亲本毒株SD-2017和传统疫苗株Bartha-K61进行了差异表达蛋白质的比较。通过生物信息学分析,本研究发现了多个与PRV影响细胞凋亡、免疫反应和自噬等相关功能的蛋白。上述结果为了解PRV致病和免疫机制,以及针对变异株探讨新的抗病毒策略奠定了理论基础。
猜你喜欢
肝博士(2022年3期)2022-06-30
音乐探索(2022年2期)2022-05-30
海外星云(2021年9期)2021-10-14
小天使·一年级语数英综合(2019年8期)2019-08-27
小学科学(学生版)(2018年7期)2018-08-13
中成药(2017年8期)2017-11-22
中国现代医学杂志(2015年26期)2015-12-23
医学研究杂志(2015年7期)2015-06-22
西安交通大学学报(医学版)(2015年2期)2015-02-28
郑州大学学报(医学版)(2015年2期)2015-02-27
