
表现为紫癜及大疱的嗜酸性肉芽肿性多血管炎一例
2023-07-06 09:45肖银玲李昕雨郭淑兰王玉坤于晓静
中国麻风皮肤病杂志 2023年7期
肖银玲 李昕雨 郭淑兰 王玉坤 于晓静
山东大学齐鲁医院皮肤科,山东济南,250012
嗜酸性肉芽肿性多血管炎(eosinophilic granulomatosis with polyangiitis,EGPA)是一种自身免疫性疾病,特征表现为成人期发病的哮喘、外周血及组织嗜酸粒细胞增多及小血管炎[1]。其特征性的病理表现为嗜酸粒细胞浸润,坏死性血管炎伴血管外肉芽肿[2]。EGPA可累及全身多个系统和器官,临床表现多样,皮肤受累很常见。现将我科诊治的一例嗜酸性肉芽肿性多血管炎患者报道如下。
临床资料患者,男,56岁,棉绒加工厂工人。2021年3月31日因双小腿皮肤出现红斑、水疱及大疱伴瘙痒10天于我科就诊。10天前患者无明显诱因双小腿出现红斑、水疱及大疱,轻度瘙痒,当地医院诊断为虫咬皮炎,并应用抗组胺药物治疗,效果不佳。既往类风湿关节炎、鼻窦炎及过敏性鼻炎病史10年。3个月前出现咳嗽、咳痰、气喘,外院胸部CT检查无明显异常,于2021年2月8日就诊于我院呼吸内科,行肺功能检查显示重度阻塞通气功能障碍,舒张试验阳性,诊断为支气管哮喘,给予孟鲁司特钠及沙美特罗替卡松治疗,肺功能恢复正常,症状缓解。
体格检查:系统检查未见明显异常。皮肤科检查:双小腿皮肤对称分布红色斑疹及斑片,压之不褪色,足踝处散在分布多个大小不等的张力性水疱及大疱,尼氏征阴性(图1)。

图1 双小腿分布红色斑疹及斑片,足踝处分布大小不等的张力性水疱及大疱,尼氏征阴性
实验室检查:白细胞计数(WBC):13.41×109/L,嗜酸粒细胞比率38.10%,嗜酸粒细胞计数5.11×109/L;红细胞沉降率88 mm/h,超敏C反应蛋白24.87 mg/L;类风湿因子>300 IU/mL,抗CCP抗体、GPI检测正常;尿常规、抗核抗体谱、核周型抗中性粒细胞胞质抗体(p-ANCA)及胞质型抗中性粒细胞胞质抗体(c-ANCA)均无异常,酶联免疫吸附试验(ELISA)显示血清抗BP180抗体、抗BP230抗体均为阴性。皮肤组织病理示(小腿):表皮内陈旧性水疱,真皮乳头高度水肿,真皮全层及皮下脂肪弥漫嗜酸粒细胞浸润(图2a),部分血管壁见嗜酸粒细胞浸润及纤维素样变性(图2b),血管外见上皮样细胞、多核巨细胞及肉芽肿(图2c)。直接免疫荧光(小腿):基底膜带C3颗粒状沉积(+)、IgG(-)、IgA(-)、IgM(-),真皮血管壁C3颗粒状沉积(+)、血管壁IgG(-)、IgA(-)和IgM(-)(图3)。
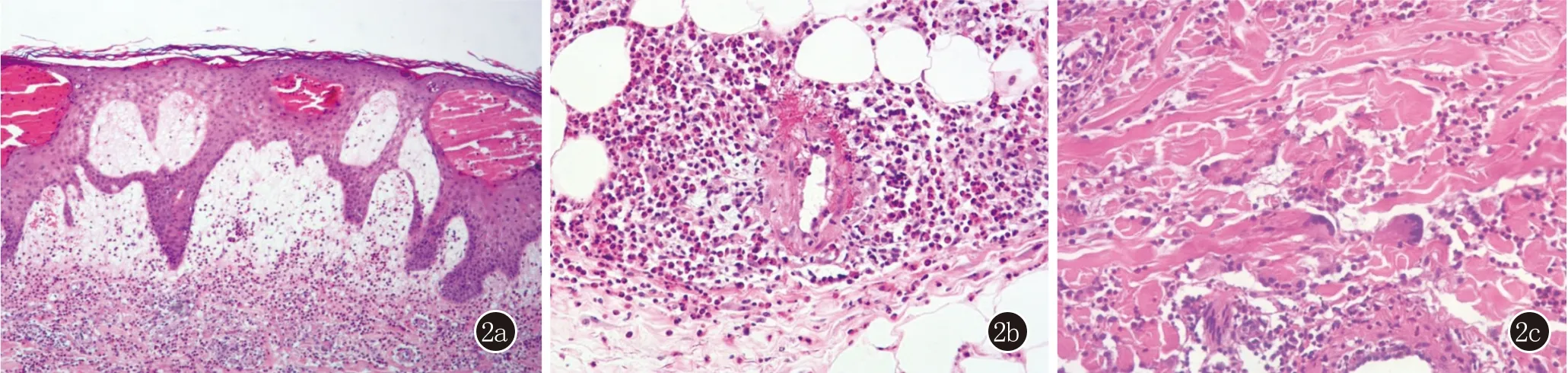
图2 2a:表皮内陈旧性水疱,真皮乳头高度水肿,真皮弥漫嗜酸粒细胞浸润(HE,×100);2b:部分血管壁见嗜酸粒细胞浸润及纤维素样变性(HE,×400);2c:胶原纤维肿胀,真皮全层散在多核巨细胞及肉芽肿(HE,×400)

图3 基底膜带C3颗粒状沉积(+),真皮浅中层血管壁C3颗粒状沉积(+)
诊断:嗜酸性肉芽肿性多血管炎。治疗:口服泼尼松25 mg/d及外用卤米松乳膏治疗。2周后紫癜样皮疹消退,水疱干涸结痂(图4),1个月后电话随访水疱痂皮脱落,无新发皮疹,糖皮质激素逐渐减量至停药,随访一年,皮疹未见复发。
讨论EGPA曾称为Churg-Strauss综合征,是一种罕见的ANCA相关性血管炎[2]。EGPA的病因尚不完全清楚,过敏原、感染、疫苗接种和药物可能是该病的触发因素[2]。
本例患者有哮喘及慢性鼻窦炎病史,外周血嗜酸粒细胞明显增多,皮肤组织病理示真皮全层嗜酸粒细胞浸润,坏死性小血管炎及血管外肉芽肿,符合1990年美国风湿病学会(ACR)提出的EGPA的分类标准[3],因此诊断为EGPA。
EGPA自然病程可分为前驱期、组织嗜酸粒细胞浸润期和血管炎期,皮肤损伤是EGPA血管炎期的一个突出表现[2]。40%~81%的EGPA患者存在皮肤表现,皮肤表现多种多样,常表现为可触及的紫癜、荨麻疹样皮疹及皮肤及皮下结节,水疱及大疱是其少见的皮肤表现[4]。在本病例中,患者表现为紧张性水疱及大疱这一少见的皮肤表现,皮肤水疱及大疱可能是由活化的嗜酸粒细胞释放细胞毒性颗粒蛋白诱导形成[5]。此外,虽然患者既往曾出现双手指间关节麻木伴疼痛,但血清中仅类风湿因子阳性,抗CCP抗体、GPI检测正常,不能明确诊断类风湿关节炎,因为在EGPA的前驱期患者也可以出现关节疼痛[2]。
虽然EGPA是一种ANCA相关的血管炎,但仅30%~40%的患者ANCA阳性,以p-ANCA为主[1]。ANCA阳性与阴性的患者在临床表型上存在差异,ANCA阳性患者肾脏受累、周围神经系统病变以及肺泡出血的发生率较高,而ANCA阴性患者发生由组织嗜酸粒细胞浸润引起的心脏受累、肺浸润和胃肠道受累的风险高[1]。本例患者ANCA阴性,目前仅皮肤组织中有大量的嗜酸粒细胞浸润。
本例患者皮损表现为紫癜样皮疹及张力性水疱、大疱,需要与以下两种疾病相鉴别:(1)过敏性紫癜:常见于儿童,成人也可发生。皮肤表现为可触及的紫癜,组织病理表现为皮肤小血管的白细胞碎裂性血管炎,无肉芽肿形成及大量嗜酸粒细胞浸润,直接免疫荧光示血管壁IgA沉积[6]。(2)大疱性类天疱疮:典型临床表现是皮肤红斑或正常皮肤上出现张力性水疱,伴皮肤瘙痒;组织病理学表现为表皮下水疱并伴有嗜酸粒细胞为主的炎细胞浸润;直接免疫荧光示IgG和/或C3在基底膜带线状沉积;ELISA检测患者血清中抗BP180和/或BP230抗体水平升高[7]。
EGPA治疗前应根据2011年修订的五因素评分(FFS)评估预后,分值越高,预后越差[3]。没有预后不良因素(FFS=0)的患者可单用糖皮质激素治疗,而预后较差的患者(FFS≥1)应采用糖皮质激素联合免疫抑制剂治疗[3]。生物制剂也是一种有前景的治疗选择,其中美泊利珠单抗已批准用于治疗EGPA[1]。该患者56岁,无心脏、肾脏及胃肠道受累的临床表现,FFS=0分,单用糖皮质激素治疗后症状明显改善。
对于有哮喘、鼻窦炎病史患者出现皮肤血管损害及外周血嗜酸粒细胞比例明显增高时需高度怀疑EGPA,当考虑到本病时应及时行组织病理学、影像学及实验室检查明确诊断。早期诊断、早期治疗可防止疾病进展,有效改善患者预后。
猜你喜欢
人人健康(2022年16期)2022-11-21
现代医院(2022年6期)2022-08-26
天津医科大学学报(2021年4期)2021-08-21
复旦学报(医学版)(2021年2期)2021-04-09
中国现代医药杂志(2020年10期)2020-12-14
河北医学(2016年5期)2016-12-01
中华老年多器官疾病杂志(2016年8期)2016-05-14
结核与肺部疾病杂志(2015年3期)2015-07-18
医学研究杂志(2015年4期)2015-06-10
中国当代医药(2015年21期)2015-03-01
