
丙酮酸脱氢酶复合物缺乏症1家系2例临床及遗传特点
2023-06-15 02:14:54刘嘉琪阮小芹张刚李杨
南京医科大学学报(自然科学版) 2023年6期
刘嘉琪,阮小芹,张刚,李杨
南京医科大学附属儿童医院神经内科,江苏 南京 210008
丙酮酸脱氢酶复合物(pyruvate dehydrogenase complex,PDHC)是一种分布于线粒体基质中的多酶复合体,通过催化丙酮酸的氧化脱羧反应生成乙酰辅酶A,有效连接糖酵解和三羧酸循环[1]。原核生物和真核生物的PDHC 含有6 种酶,丙酮酸脱羧酶(pyruvate decarboxylase,简称E1)、二氢硫辛酰胺乙酰转移酶(dihydrolipoamide acetyltransferase,简称E2)和二氢硫辛酰胺脱氢酶(dihydrolipoamide dehydrogenase,简称E3)是组成PDHC 最关键的3 种酶,其中E1 是由两个α和两个β亚基组成的异四聚体[2]。丙酮酸脱氢酶复合物缺乏症(pyruvate dehydrogenase complex deficiency,PDCD)是一种罕见的线粒体能量代谢障碍遗传性疾病,起病隐匿,约84%的患者为定位于Xp22.1~22.2区域的PDHA1基因发生突变[3],可导致E1的α亚基(E1-α)功能缺陷,出现高乳酸血症、进行性神经肌肉损害、发育迟缓等多种临床症状[4]。现分析1家系2例诊断为PDHA1基因突变导致的PDCD女童的临床资料及基因分析结果,为其临床诊断、遗传咨询及后期治疗提供依据,并以期提高临床医师对本病的认识。
1 病例资料
例1:患儿,女,9 岁3 个月,2022 年1 月因“咳嗽2周,行走不稳1周”入院,以呼吸道感染症状起病,病初有气促、喘息及呼吸困难,曾外院机械通气2 d后顺利撤机,病程中有头痛、口齿不清、行走不稳症状,无明显抽搐,四肢肌力下降。患儿系G1P1,早产(33 周)剖宫产。患儿既往学习成绩处于班级中下等,语速较慢,运动协调能力差,既往行走姿势异常、高足弓。父母身体健康,非近亲结婚,否认家族遗传病史及传染病史。入院查体:神志清,精神反应欠佳,面色正常,呼吸促,可见轻度吸凹,双侧瞳孔等大等圆,对光反射灵敏,咽部稍充血,颈软,双肺呼吸音粗,可闻及少许痰鸣音及喘鸣音,心腹未见明显异常,四肢肌力Ⅳ级,双下肢肌张力减低,脑膜刺激征阴性,双侧巴氏征阴性,双侧腱反射可引出。辅助检查:血常规、生化全套、凝血、血沉、自身抗体、肿瘤4 项、微量元素、甲状腺功能基本正常;EB病毒、单纯疱疹病毒、流感病毒、肺炎支原体等病原学检查阴性;脑脊液白细胞1 个/μL,淋巴细胞70%,单核细胞30%,脑脊液葡萄糖及蛋白正常;血清及脑脊液中枢神经系统脱髓鞘疾病自身抗体:水通道蛋白4(aquaporin4,AQP4)抗体IgG、髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoprotein,MOG)抗体IgG、髓鞘碱性蛋白(myelin basic protein,MBP)抗体IgG均阴性;血尿遗传代谢筛查:血丙氨酸增高(923.21 μmol/L),血乳酸增高(6.6 mmol/L),尿乳酸及丙酮酸增高;眼底检查正常;WISC-R 学龄儿童智力测验量表:常识1分、填图5分、类同2分、积木6分,测试总量表14分,智商IQ 58分(轻度弱智),配合良好;视频脑电图提示异常儿童脑电图,清醒期左侧前头部尖波、慢波发放;肌电图提示轻度神经源性损害肌电改变;视觉听觉诱发电位正常;体感诱发电位正常;全脊柱MRI未见明显异常;头颅MRI(2022 年1 月4 日)提示两侧小脑、基底节区及侧脑室旁多发异常信号(图1)。入院诊断:病毒性脑炎?代谢性脑病?重症肺炎。予阿昔洛韦抗病毒、头孢地嗪抗感染、地塞米松抗炎、甘露醇脱水降颅压、维生素B1、辅酶Q10、左卡尼汀等治疗,治疗后血乳酸明显下降至2.3 mmol/L,患儿行走不稳、肌力肌张力下降等症状好转后出院。患儿出院后定期于本院康复科行康复训练治疗,并口服维生素B1、辅酶Q10、左卡尼汀及生酮饮食等治疗,运动能力较前改善,能独站,独走稍不稳、双手抓物轻度震颤、反应迟钝、言语口齿欠清。出院后定期本院神经内科复诊,复查头颅MRI(2022年8月29日)提示两侧小脑、基底节区及侧脑室旁多发异常信号较前好转,局部软化灶形成(图2)。近期随访发现患儿运动及认知能力较前改善,目前独走稳,言语较前清晰。
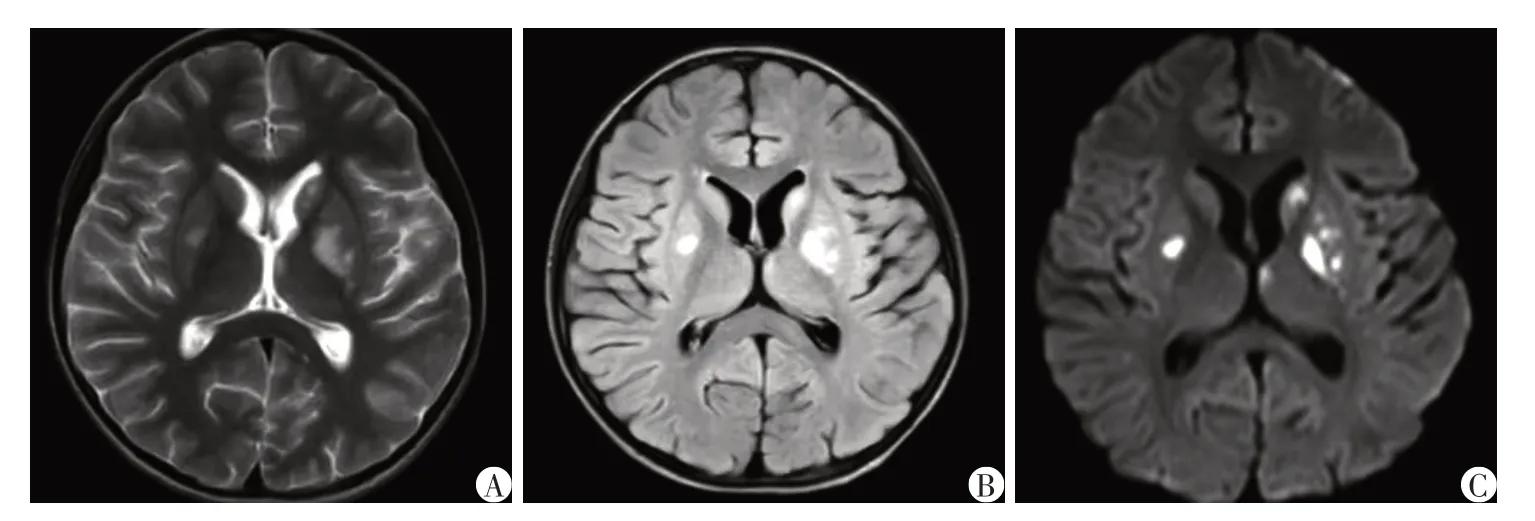
图1 例1患儿入院时头颅MRI检查图像
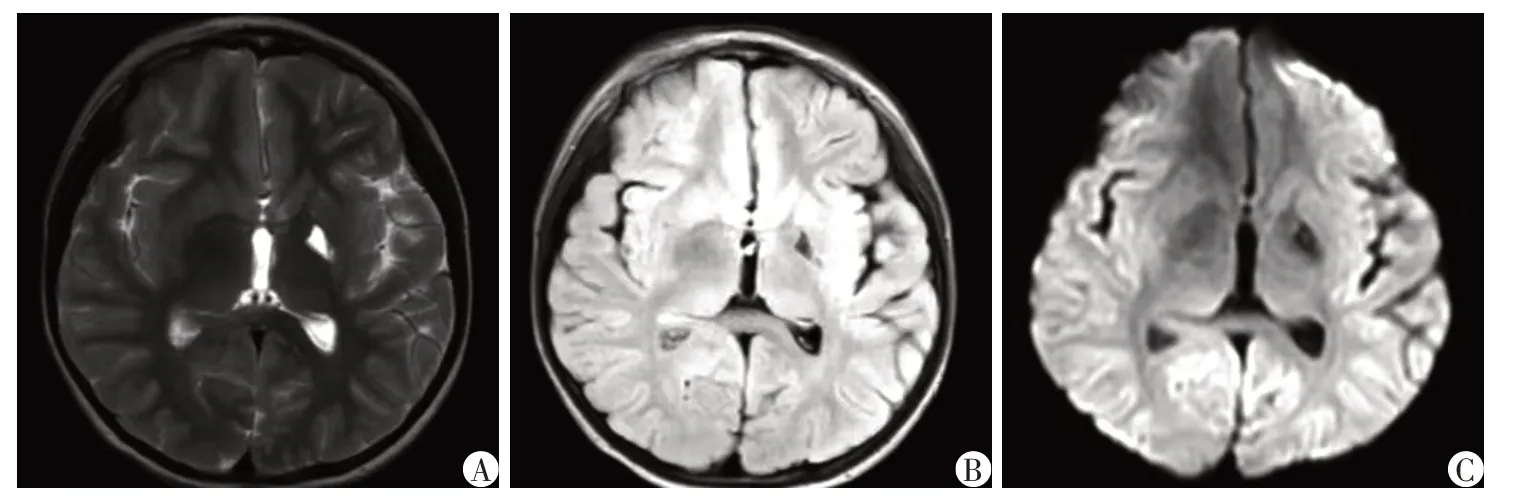
图2 例1患儿复诊时头颅MRI检查图像
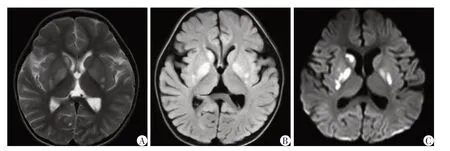
图3 例2患儿入院时头颅MRI检查图像

图4 例2患儿复诊时头颅MRI检查图像
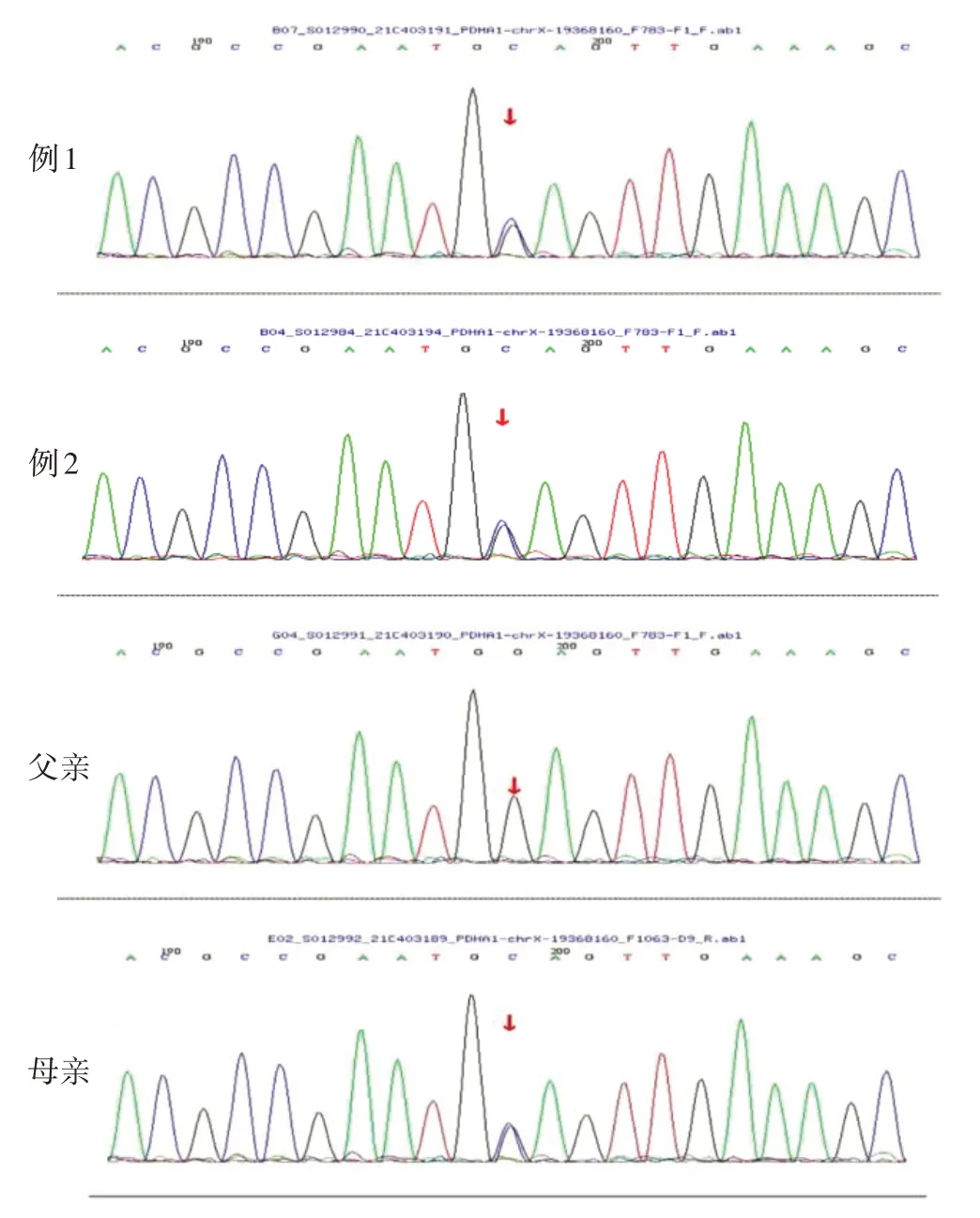
图5 患儿及其父母的基因测序图
例2:患儿,女,2岁,2022 年1 月因“咳嗽1周,间断发热伴抽搐4 d”入院。患儿与其胞姐同期以呼吸道感染症状起病,有发热、咳嗽、意识障碍、行走不稳,病程中出现多次抽搐发作,表现为意识丧失,双眼上翻,四肢抽动,无口吐白沫,无大小便失禁,持续数分钟至1 h。患儿系G2P2,足月剖宫产,新生儿期有“肺炎”病史,于当地医院住院治疗10余天后好转出院。既往生长发育落后于一般同龄儿,18月龄会走路,目前只能说叠字单词、不会说长句。父母身体健康,非近亲结婚,否认家族遗传病史及传染病史。入院查体:神志欠清,精神反应欠佳,面色正常,呼吸平稳,双侧瞳孔等大等圆,对光反射灵敏,咽部充血,颈软,双肺呼吸音粗,可闻及少许细湿啰音,心腹未见明显异常,四肢肌力Ⅳ级,肌张力减低,脑膜刺激征阴性,双侧巴氏征阴性,双侧腱反射可引出。辅助检查:血常规、生化全套、凝血、血沉、自身抗体、肿瘤4项、微量元素、甲状腺功能基本正常;EB 病毒、单纯疱疹病毒、流感病毒、肺炎支原体等病原学检查阴性;脑脊液白细胞28个/μL,中性粒细胞27%,淋巴细胞46%,单核细胞27%,脑脊液葡萄糖及蛋白正常,脑脊液二代测序阴性;血清及脑脊液中枢神经系统脱髓鞘疾病自身抗体:AQP4抗体IgG、MOG 抗体IgG、MBP 抗体IgG 均阴性;血尿遗传代谢筛查:血乳酸增高(5.0 mmol/L),尿乳酸及丙酮酸正常;眼底检查正常;DST 儿童发育筛查:DQ 51分(发育商低下)、MI 77分(智力指数可疑低下),其中运动智龄=21个月、社会适应智龄=18个月、智力智龄=27个月;视频脑电图提示异常幼儿脑电图,背景活动减慢,清醒期双侧额极1.5~2.5 Hz δ活动发放;肌电图提示轻度多发性周围神经源性损害肌电改变(运动和感觉神经轴索损害为主);视觉听觉及体感诱发电位正常;全脊柱MRI未见明显异常;头颅MRI(2022年1月6日)提示两侧基底节区及侧脑室旁多发异常信号(图3)。入院诊断:病毒性脑炎?惊厥持续状态、代谢性脑病?支气管肺炎。予阿昔洛韦抗病毒、头孢地嗪抗感染、甘露醇脱水降颅压、维生素B1、辅酶Q10、果糖二磷酸钠、左卡尼汀等治疗,治疗后血乳酸下降至3.8 mmol/L,患儿抽搐、行走不稳等症状好转后出院。患儿出院后定期于本院神经内科复诊,继续口服维生素B1、辅酶Q10、左卡尼汀及生酮饮食治疗,仍独走稍不稳,语言发育迟缓,只会说短叠词,复查头颅MRI(2022年9 月5 日)提示两侧基底节区异常信号明显好转(图4)。近期随访发现患儿运动及认知能力较前改善,独走稳,语言可以说短句。
本研究获得本院伦理委员会批准(伦理号:202210189-1),在获得患儿父母知情同意后,治疗期间抽取2例患儿及其父母外周静脉血各2 mL,分别保存于EDTA-K2抗凝管中,委托北京迈基诺医学检验所对2例患儿应用高通量测序进行全外显子组基因检测及线粒体基因组测序,从而发现可能存在的致病突变,再通过Sanger 测序对患儿及父母双方可疑致病突变位点进行验证。应用高通量测序在2例患儿的X染色体上PDHA1基因(NM_000284)的第3外显子发现一错义突变c.223G>C(p.E75Q),223号核苷酸鸟嘌呤G 变为胞嘧啶C,该变异导致第75 位氨基酸由谷氨酸变为谷氨酰胺,为可疑致病性突变,检索相关文献提示与PDHC E1-α亚单位缺陷相关,将2例患儿与其父母样本进行Sanger测序验证,发现其父亲该位点无变异,其母亲该位点杂合变异(图5),可分析出该家系遗传系谱图(图6)。查询人类基因突变数据库HGMD(Human Gene Mutation Database,http://www.hgmd.org)、Clinvar 数据库,无该位点致病性分析结果,查阅文献数据库未有该位点的相关性报道,c.223G>C为未报道过的新变异位点。同时进行跨物种氨基酸保守性分析提示该位点在长臂猿、狨猴、非洲爪蛙等多物种间高度保守(图7),说明该氨基酸可能具有重要的生物化学功能,在此区域发生变异可能会引起蛋白质结构与功能异常。采用PyMOL 软件分析对比野生型与突变型蛋白的三级结构,发现此变异可引起PDHA1基因编码蛋白结构明显改变,突变型蛋白中75位谷氨酸(Glu)突变成谷氨酰胺(Gln),突变后导致其无法通过氢键与94 位半胱氨酸(Cys)结合,故会影响其功能活性(图8)。检索HGMD数据库、PubMed及Clinvar 数据库,参考相关文献,根据美国医学遗传学与基因组学学会(the American College of Medical Genetics and Genomics,ACMG)2015 年发布的基因变异解读标准和指南进行致病性判读,c.223G>C判定为疑似致病性变异,PM1+PM2+PM5+PP3(PM1:该变异位于突变热点区域;PM2:在正常人群数据库中为低频变异;PM5:相同位置的突变在文献数据库/Clinvar 数据库已有报道,但氨基酸变化不同;PP3:生物信息学蛋白功能综合性预测软件REVEL 预测结果为有害,SIFT、PolyPhen_2、Mutation Taster、GERP+预测结果均为有害)。因此,此新变异可能为致病性变异,结合2 例患儿临床表现,确诊为PDCD。
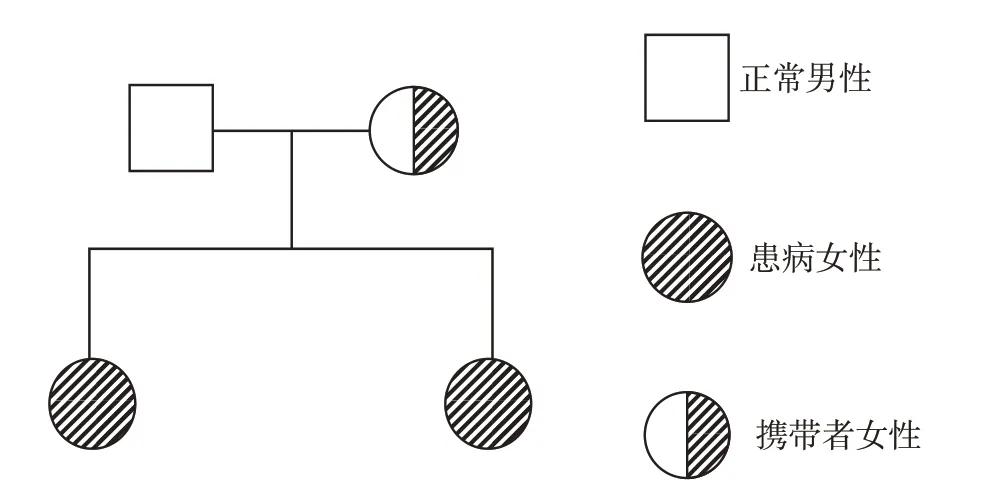
图6 患儿及其父母PDCD家族遗传系谱图

图7 氨基酸跨物种保守性分析
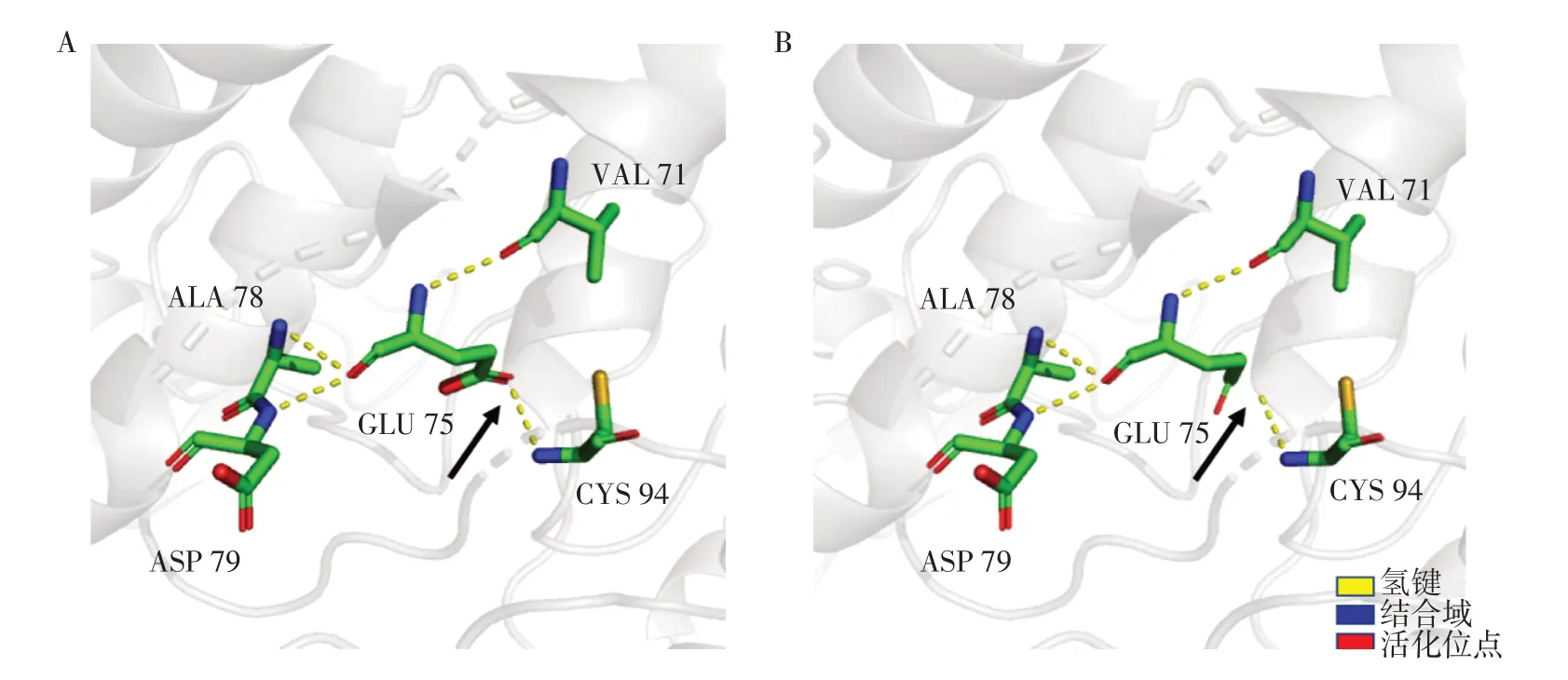
图8 蛋白质的三级结构比对
2 讨论
PDCD 最早于1970 年被报道,是一种线粒体能量代谢障碍的X 连锁遗传性疾病,最常见的临床表现包括神经系统症状综合征(先天性小头畸形、肌张力障碍、癫痫、共济失调)、脑成像异常(胼胝体发育不良、Leigh 综合征)和代谢异常(血浆丙酮酸升高、高乳酸血症或代谢性酸中毒),大多数患者均存在精神运动发育迟缓[5]。PDCD 的体征和症状在不同患者之间有显著差异,一般来说,发病年龄和临床症状谱之间存在相关性,通常在婴幼儿和儿童早期发病,病情轻重及存活时间的长短与乳酸酸中毒的严重程度密切相关,男女发病比例无明显差异,但一些严重症状对男性的影响往往比对女性的影响更大,男性病死率高于女性。PDCD 新生儿期的典型表现为严重的致死性高乳酸血症[6]、伴有呼吸急促、肌张力减退和昏迷,常伴大脑发育不全,在男性和女性中都可见。婴儿期起病患者的临床表现:严重的发育迟缓、常伴有癫痫,多见于婴儿早期发病的女性;发病于婴儿期的男性更有可能出现Leigh综合征或脑干功能障碍等。少数患者起病时症状较轻微,多见于年龄较大的儿童和青少年[7],在给予高碳水化合物饮食后出现复发性共济失调,缓慢发展为Leigh 综合征。另外,部分女性患者也可出现先天畸形表现,包括小头畸形、宽鼻梁、喇叭状鼻孔、鼻孔前翻、长人中、低位耳、手指短、四肢近端短及肛门前移等。罕见的迟发性PDCD在国外已被报道,两兄弟在40 多岁时出现运动障碍,包括非典型帕金森病、舞蹈运动、刻板动作、共济失调和构音障碍,在皮肤成纤维细胞分析试验中发现了PDHC 的缺乏[8]。
PDHC是一个多酶复合物,主要由3个功能亚基E1、E2和E3组成,其中E1是一种异四聚体,由2个α亚基和2 个β亚基组成,PDHC 的核心结构E1-α由PDHA1 基因编码[9]。PDHA1 基因是PDCD 最常见的突变基因,其位于X 染色体短臂Xp22.1~22.2上,全长约1.75 kb,含有11 个外显子,当其发生突变时使E1从丙酮酸脱羧基及剩余的乙酰基转移到E2的过程受阻,导致PDHC 失活,出现丙酮酸代谢障碍,进而出现高乳酸血症、进行性神经肌肉损害、发育迟缓等一系列临床症状。本文2例同胞患儿分别因呼吸道症状伴运动障碍、呼吸道症状伴癫痫发作就诊,查体均发现肌张力障碍,血乳酸明显增高,例1患儿尿液有机酸分析提示丙酮酸明显增高,血丙氨酸增高,头颅核磁检查提示两侧小脑、基底节区及侧脑室旁多发异常信号。例2 患儿血乳酸增高,头颅核磁检查提示两侧基底节区及侧脑室旁多发异常信号。2 例患儿PDHA1 基因均检出c.223G>C 错义突变,生物信息学预测分析为疑似致病突变,证实为PDHC E1-α亚单位缺陷,其母亲该位点杂合变异,其父亲该位点无变异。患儿母亲既往体健,无明显相关症状,考虑与女性X 染色体的随机失活模式相关,导致不同患者间的PDHC 残存酶活性存在差异,故PDHA1基因突变的女性可能症状不明显或无症状[10]。多数PDHA1基因为自发突变,仅少数突变来源于遗传,本研究2例患儿PDHA1基因检测均发现与其母亲存在同一位点相同突变。通过查阅HGMD数据库,目前已报道的PDHA1基因突变类型超过210种,包括错义突变、无义突变、剪接突变、插入突变和缺失突变等,其中错义突变为主要方式。本研究2例患儿为c.223G>C错义突变,223号核苷酸由鸟嘌呤G 变为胞嘧啶C(c.223G>C),导致第75 位氨基酸由谷氨酸变为谷氨酰胺(p.E75Q),REVEL、Polyhen_2、SIFT、Mutation Taster、GERP+软件均预测此突变可能具有致病性。目前尚未有该位点的相关文献报道,Clinvar 数据库尚无该位点致病性分析结果。通过检索PubMed数据库、万方数据库及中国知识基础设施工程(CNKI)数据库,以“丙酮酸脱氢酶复合物缺乏症”、“PDCD”、“PDHA1”等为关键词查找中国人PDCD 病例,自建库至2022 年12月,共检索出16 种变异类型,包括本例研究,共有17种变异类型,其中错义突变14 种(R72C、E75Q、R126C、A136V、E143K、A169V、N202S、F205L、C214T、R263G、R270G、G278E、R378H、R378C),插 入突变3 种(c.1111_1158dup48bp,p.S371_F386dup16;c.1243_1257dup15,p.V415-N419dup;c.1159_1162dupAAGT,p.S388Terfs,图9)。本例c.223G>C(p.E75Q)突变为中国人群PDHA1 基因变异数据库增添了新的研究样本。
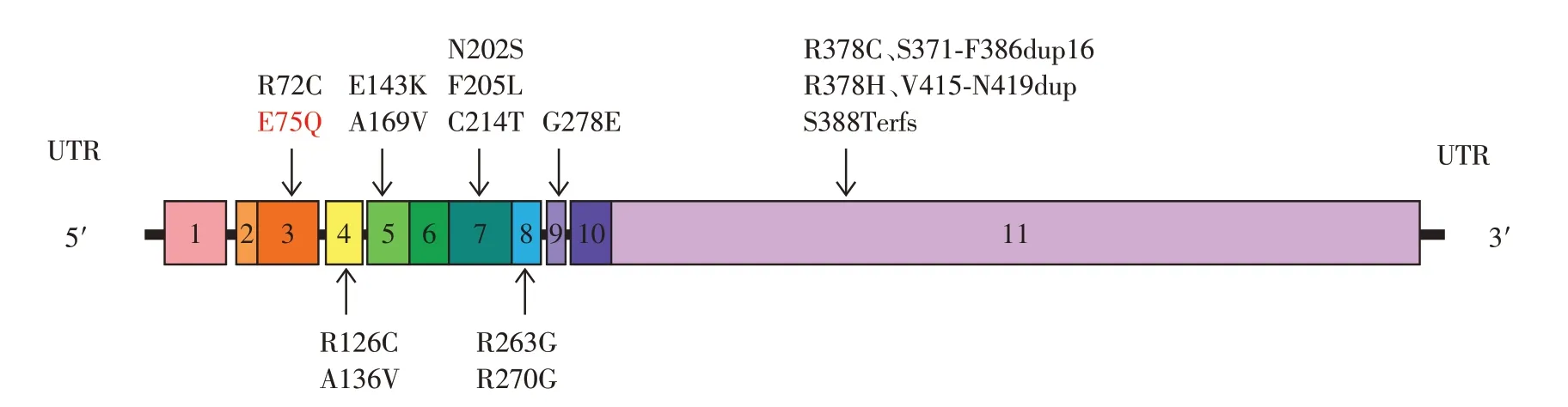
图9 中国人PDCD病例变异类型
PDCD现尚无根治方法,只能对症治疗,预后较差,疾病早期给予维生素B1 及辅酶Q10 等治疗,有助于改善神经系统临床症状及预后。生酮饮食是原发性PDCD 患者的金标准疗法[11],生酮饮食是一种高脂肪、低碳水化合物、低蛋白饮食,模仿长期禁食的代谢状态。在PDCD 患者中,糖酵解最终产物丙酮酸没有通过三羧酸循环得到最佳代谢,导致乳酸增加,并通过线粒体呼吸链损害三磷酸腺苷(ATP)酶的生产。在碳水化合物剥夺期间,细胞能量不再来自糖酵解,而是来自脂肪酸的分解。脂肪酸分解产生的酮体(β-羟基丁酸、乙酰乙酸和丙酮)可以通过血脑屏障进入中枢神经系统为大脑提供能量[12]。生酮饮食已被证明在控制癫痫发作,改善共济失调、乳酸性酸中毒和睡眠障碍方面有效,Sofou等[5]研究还表明,生酮饮食能改善语言、社交、认知和运动发育等。施行生酮饮食疗法后应长期随访及健康教育,提高遵医行为,定期监测血酮、血糖、尿酮等,协助解决患儿家庭在生酮饮食治疗期间遇到的难题。另外,国外有研究发现PDCD 的潜在治疗方法包括二氯乙酸盐和苯基丁酸盐[13-14],该两种药物既往用于其他疾病,如癌症和枫糖尿症等。二氯乙酸盐可降低血乳酸水平,苯基丁酸盐可通过抑制丙酮酸脱氢酶激酶(pyruvate dehydrogenase kinase,PDK)活性干扰PDHC 和其激酶之间的结合,使PDHC保持活性,故可能成为生酮饮食的替代治疗,但其不良反应尚不完全明确,如周围神经毒性等,由于这种先天性代谢病是一种早期疾病,因此需要详细了解对儿童的潜在不良反应,需要进一步进行细胞培养、动物模型和临床试验的研究,以充分了解PDCD 及其治疗方法。本研究中2例患儿均已予维生素B1、辅酶Q10、左卡尼汀、生酮饮食等治疗,对延缓病情、改善症状有一定作用,使得生存质量得到明显提高。
综上,PDHA1 基因突变致PDCD 为罕见遗传性疾病,其临床症状复杂多样,临床工作中极易误诊、漏诊,临床诊断困难且预后不佳,易在新生儿期或儿童期死亡,对临床上高乳酸血症及有神经肌肉系统病变的患儿应警惕此病,故应注意进行详细的临床检查,积极完善基因检测,定期复查头颅核磁等。另外,对先证者家庭进行遗传咨询及产前诊断亦非常必要。该病目前尚无根治方法,以对症治疗为主,早期诊断、及早治疗对改善预后极为重要,后续应收集及分析更多该类病例,有助于进一步了解PDCD及进行干预治疗。
猜你喜欢
工业微生物(2024年1期)2024-02-29 07:36:50
生物加工过程(2023年6期)2023-12-11 03:27:52
家庭医药·快乐养生(2023年5期)2023-05-19 20:53:22
牡丹江医学院学报(2021年5期)2021-12-05 08:01:51
中华养生保健(2020年8期)2021-01-14 01:13:58
心肺血管病杂志(2020年3期)2021-01-14 00:42:38
——生酮饮食
中老年保健(2020年12期)2020-12-09 11:46:36
科学生活(2018年12期)2018-12-25 12:20:56
腹腔镜外科杂志(2016年12期)2016-06-01 12:10:09
河北科技大学学报(2015年6期)2015-03-11 16:16:51
