
电压门控钠离子通道在难治性癫痫中的作用机制研究进展
2023-06-15 02:14:54张乔孟帆周其冈
南京医科大学学报(自然科学版) 2023年6期
张乔,孟帆,周其冈
南京医科大学药学院,江苏 南京 211166
难治性癫痫(treatment-resistant epilepsy,TRE),又称耐药性癫痫(drug-resistant epilepsy,DRE),其药物治疗是目前临床医学的重大难题之一。除了手术治疗之外,尚无较好治疗方案,导致患者过早死亡、受伤、心理社会功能障碍和生活质量降低的风险增加[1]。在过去20 年里,大量新抗癫痫药物(antiepileptic drug,AED)被开发应用于癫痫治疗,提高了易用性和耐受性,但这些AED均不能显著预防或逆转TRE[2]。
癫痫发病机制主要是大脑神经元异常同步化放电,并向周围组织扩散。离子通道在神经元异常放电过程中发挥重要作用。其中,电压门控钠离子通道(voltage-gated sodium channel,VGSC)是最常见的AED靶点,它的结构或功能改变可能与TRE形成密切相关[3]。VGSC 在兴奋性神经元和抑制性神经元中均有表达,在动作电位的产生与传播过程中发挥关键作用。动作电位是神经元电生理最基本的活动,离子通道结构和功能异常导致动作电位节律异常,诱发严重临床症状,包括癫痫发作、智力低下、行为异常和运动障碍等。VGSC家族亚型SCN1A(编码Nav1.1)、SCN2A(编 码Nav1.2)和SCN8A(编 码Nav1.6)基因共同占脑钠通道转录物95%以上,和目前已知绝大多数脑钠通道病因相关[4]。在一项调查中,8 565 例癫痫和神经发育障碍患者中有5%携带了这些基因中的一种变异[5]。本文就VGSC 在TRE发病机制中的研究进展进行综述。
1 TRE
1.1 概念
癫痫是大脑组织局部神经元异常放电,并向周围组织扩散,导致神经系统紊乱的慢性疾病。该病的特点是反复发作,并伴有认知、心理和社会后果。2019 年全球约有7 000 万癫痫患者,其中90%以上在低、中等收入国家[6]。2021 年中国最新流行病学资料显示,我国约有1 000万癫痫患者,但仅有1/3的患者可以得到适当的治疗[7]。
TRE 一直是临床上的棘手难题,国际抗癫痫联盟将其定义为根据癫痫发作类型,合理选择并正确使用至少2 种耐受性好的AED 单药或联合使用后,患者无发作的持续时间未达到治疗前最长发作间隔的3倍或者1年[8]。
1.2 TRE发病机制
TRE 的发病机制是多因素和可变的,可能包括遗传和环境因素等[9]。目前关于TRE发病机制的研究仍不透彻,主要有以下几个方面的假说。
1.2.1 海马硬化
海马硬化是TRE 最常见的病因[10],临床上主要通过联合AED和手术切除进行治疗,但AED治疗效果不佳。海马硬化的特征是神经元不同程度丢失,尤其是CA1 和CA3区域的锥体细胞严重丧失,反应性胶质细胞增生,颗粒细胞分布异常和苔藓纤维出芽,其中苔藓纤维出芽会导致海马内形成异常的兴奋性环路,这是导致癫痫反复发作的重要病理学基础[11]。
1.2.2 离子通道
离子通道突变会导致遗传性癫痫的发生,而遗传性癫痫多为难治性的[12]。离子通道是各种阴离子、阳离子跨膜被动转运的通道,参与调节静息膜电位、产生动作电位,还参与调节中枢神经系统,为兴奋及抑制等提供基础。离子通道在癫痫的阵发性异常放电过程中发挥了重要作用[13]。
1.2.3 炎症
炎症与TRE的发生发展息息相关[14],目前有研究发现炎症可通过激活细胞内信号通路,使脑血管内皮细胞及血脑屏障胶质细胞中转运体异常表达和功能障碍,诱导TRE的发生[15]。也有其他研究表明,促炎细胞因子白介素(interleukin,IL)-1β可能通过抑制星形胶质细胞对谷氨酸的再摄取或增强N-甲基-D-天冬氨酸和α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体功能,调节谷氨酸能系统活性,导致癫痫反复发作,进而参与TRE形成[16]。炎症与TRE的关系已被大量证实,但是发病机制仍未阐明。
1.2.4 线粒体疾病
线粒体疾病是最常见的遗传代谢紊乱性疾病,以能量生成和调节障碍为特征。线粒体疾病也是神经遗传性疾病之一,它的常见症状之一是癫痫发作,但是常用的AED治疗对于绝大多数病例无效[17],超过90%的线粒体相关癫痫的儿童患者是药物难治性的[18]。线粒体疾病参与TRE 发生发展是不容置疑的,但是两者之间的关系以及作用机制还不明确。目前研究表明,线粒体相关癫痫患者的星形胶质细胞在神经元受损区域增生。星形胶质细胞中谷氨酰胺合成酶缺乏不仅可导致线粒体疾病发生,还可引起γ-氨基丁酸(γ-aminobutyric acid,GABA)-谷氨酸-谷氨酰胺循环的下调,造成脑组织中GABA降低,引起癫痫发作[19]。
1.3 TRE耐药机制
虽然接受颞叶癫痫病灶切除手术的癫痫患者的组织可用于基础研究,使得阐明局灶性癫痫耐药的一些潜在机制成为可能,但是全身性癫痫产生耐药性的机制仍未阐明,对这些潜在的细胞和分子机制进行探索,很可能为发展新的药物治疗策略提供重要方向。癫痫的耐药机制尚不明确,可能是多因素的,而研究较多的是以下几种假说(图1)。

图1 TRE的耐药机制假说Figure 1 Hypothesis of drug resistance mechanism in TRE
1.3.1 药物转运体假说
癫痫的耐药性有一个重要特征,即它对多种作用机制不同的AED有着同样的耐药性,说明TRE的耐药机制可能是非特异性的。在TRE 患者切除病灶的毛细血管内皮中,发现外排转运蛋白表达显著上调,这个结果支持外排转运蛋白在癫痫耐药发病机制中发挥作用的假设[20]。这些蛋白在反应性星形胶质细胞和神经元中有新的表达,可能形成了“第二防御”机制[21]。脑内过多表达药物转运蛋白,将AED从致痫灶中过度外排,从而影响药物在致痫组织中的浓度,导致药物难以发挥疗效[22]。
例如,P 糖蛋白(P-glycoprotein,P-gp)由多药耐药基因1 编码,是血脑屏障上的主要药物外排转运蛋白,可运输结构多样且不相关的疏水性和两亲水性化合物,并已被证明可运输苯妥英钠、苯巴比妥、拉莫三嗪、托吡酯和左乙拉西坦等AED。近年研究表明,P-gp 过表达是TRE 发病的主要机制之一[23]。在TRE患者的致痫组织以及几种啮齿动物癫痫模型中,检测到P-gp的表达水平升高。并且癫痫持续发作诱导的谷氨酸信号释放,导致P-gp在脑毛细血管中的表达增加[24]。P-gp通过协调AED结合位点的构象变化和三磷酸腺苷的结合、水解及其产物释放,以能量依赖方式限制血脑屏障对AED的渗透,使AED不能进入脑内发挥有效抗癫痫作用[25]。
1.3.2 药物靶点学说
药物发挥作用需要与靶点结合,当结合靶点的分子结构或功能发生变化时,药物治疗敏感性降低。AED 作用靶点主要有离子通道,如Na+通道、Ca2+通道、K+通道,及递质作用受体,如GABA受体[26]。其中VGSC是最常见的AED靶点,代表药物有苯妥英钠、卡马西平、拉莫三嗪等。临床研究表明,对卡马西平耐药的颞叶癫痫患者与不耐药患者的海马相比,齿状回(dentate gyrus,DG)颗粒细胞功能依赖性钠离子通道阻断作用完全丧失[27]。在耐药的颞叶癫痫患者中也观察到GABAA受体表达的改变,但这种改变是否会导致患者对作用于该受体的AED的敏感性降低,目前尚不清楚[28]。
1.3.3 神经网络假说
癫痫反复发作导致脑内神经元突触可塑性改变,以及神经网络中信号分子诱导神经元轴突的异常生长,包括轴突发芽、突触重组、神经发生和胶质增生等。这些改变诱发的神经网络退化和重塑,抑制了内源性抗癫痫系统,并阻止AED进入神经元靶点,从而导致TRE。有研究人员对TRE患者和健康人在静息状态下的功能性磁共振成像数据进行分析发现,患者的神经网络连接功能有所下降,表明癫痫发作可能干扰大脑神经网络的相互作用,并进一步影响神经元的信息交流[29]。虽然该假说并不能对所有的TRE耐药机制进行解释,但可以提供一种新的方向。
2 VGSC与TRE
2.1 VGSC结构与功能
VGSC 由1 个230~270 kDa 的α亚基和1 个或多个33~36 kDa 的β亚基组成。α亚基包括4 个同源结构域,每个结构域包含6 个高序列保守的跨膜片段[30]。β-亚基影响α-亚基的转运和电生理特性,但自身不具有通道活性。人类α亚基的基因家族包含10 个基因,其中SCN1A、SCN2A 和SCN8A 基因在中枢神经系统的神经元中表达量较高,占95%以上[4];其编码的钠通道蛋白分布于神经元轴突、树突和胞体的细胞膜上[31]。钠离子通道基因是人类基因组中高度进化保守的基因之一,并保留了许多与无脊椎动物和原核生物钠通道序列相同的区域。偏离正常通道功能会导致严重的临床后果,包括癫痫发作、智力残疾、行为异常和运动障碍。为了响应跨膜电势的去极化位移,VGSC 带正电的跨膜段的构象从封闭通道状态转变为开放通道状态,允许Na+的涌入和动作电位的启动;随后通道孔被失活门阻塞,通道在几毫秒内发生快速失活,进入非活性构象,因而Na+的流入被限制在短暂的间隔内。当失活门恢复到其静止位置,VGSC 从失活中恢复并维持在稳定的闭合构象,这一进程的改变是许多钠通道突变机制的基础[32]。
2.2 VGSC与TRE
2.2.1 VGSC基因突变导致TRE
编码VGSC的基因突变会导致以药物难治性类型为主的遗传性癫痫。其中,近几年的研究发现SCN1A、SCN2A和SCN8A基因突变引发了多种临床疾病[33](表1)。其变异主要分为离子通道功能缺失型(loss-of-function,LOF)与离子通道功能获得型(gain-of-function,GOF)。钠离子功能减弱,其癫痫发生较晚,但能引起更重的癫痫表型,且使用钠离子通道阻滞剂会加重癫痫发作[34]。
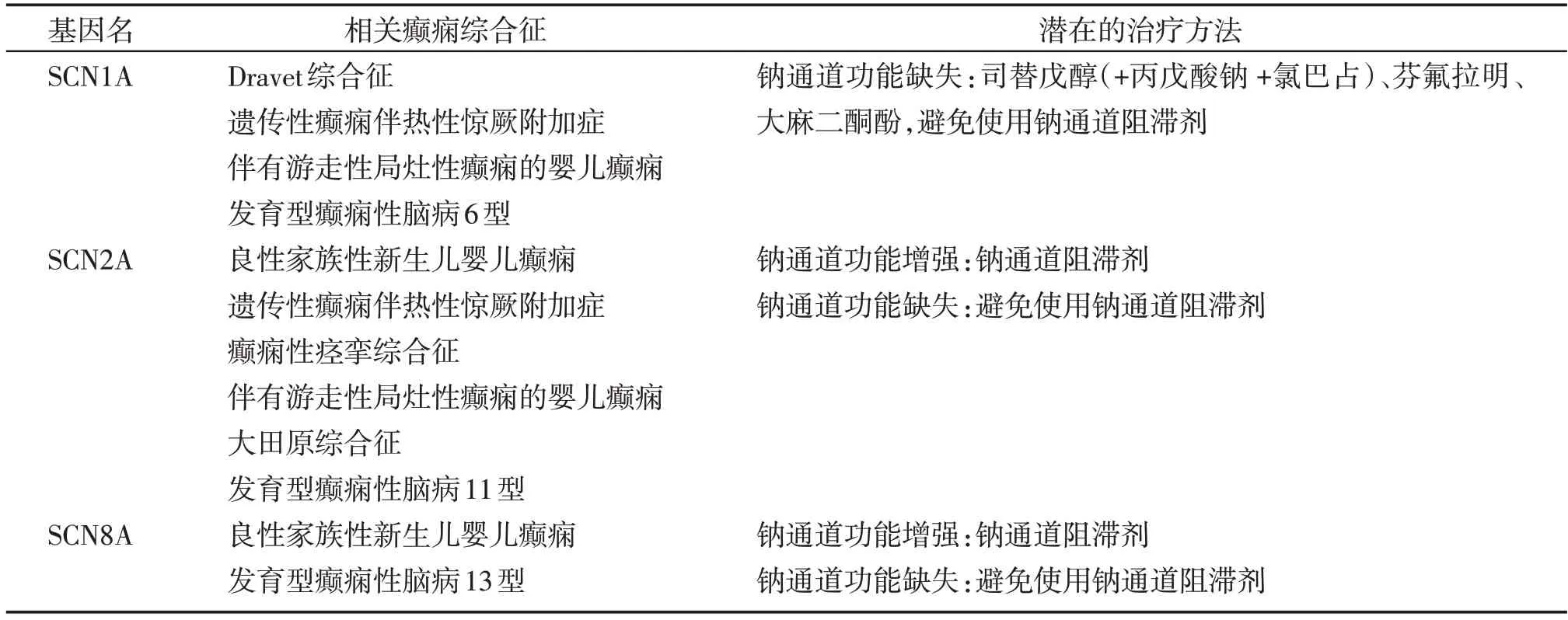
表1 VGSC基因型-表型关系以及潜在的精准治疗方法Table 1 Genotype-phenotype correlation of VGSC and potential precision therapies
SCN1A 编码的Nav1.1 主要分布于抑制性神经元,在小脑浦肯野纤维、海马抑制性GABA 能神经元、下丘脑及大脑皮质的中间神经元均可检测到一定程度的表达[35]。在细胞水平,Nav1.1 主要位于细胞体和轴突起始段轴突起始节(axon initial segment,AIS)[36]。SCN1A 基因突变是导致遗传性癫痫综合征中最常见的突变靶点,可导致Nav1.1的C端、跨膜段、胞内段和N 端都出现突变[37]。Dravet 综合征(Dravet syndrome,DS),又称婴儿严重肌阵挛性癫痫(severe myoclonic epilepsy in infancy,SEMI),其中85%的DS 患者存在SCN1A 基因突变[38],与DS 相关的基因突变类型主要是框移突变、无义突变以及错义突变,均导致Nav1.1功能障碍。2000年首次在遗传性全面性癫痫伴热性惊厥附加症(generalized epilepsy with febrile seizures plus,GEFS+)中发现了SCN1A 基因突变[39],占GEFS+患者的5%~10%[40]。与DS患者的基因突变不同,几乎所有与GEFS+相关的SCN1A基因突变类型都是错义突变,且主要集中在Nav1.1的胞内段。DS的临床表现较GEFS+更加严重,对于AED的治疗反应不佳,且卡马西平、奥卡西平、拉莫三嗪等钠离子通道阻断剂会加重发作,不建议使用这些药物[38]。
SCN2A编码的Nav1.2蛋白,于发育早期在有髓神经纤维的AIS 和Ranvier 节点中表达,到1~2 岁时逐渐被Nav1.6 取代。成人时期,Nav1.2 主要在兴奋性神经元AIS及未髓鞘化轴突部位表达[41]。SCN2A基因突变会引起Nav1.2表达和功能变化,造成神经元异常放电,从而导致多种癫痫的发生,包括轻度的自限性家族性新生儿癫痫、大田原综合征、婴儿癫痫伴游走性局灶性发作、West 综合征及Lennox-Gastaut 综合征等[33]。SCN2A 基因突变主要见于癫痫罕见基因突变,与TRE的发病有密切关系。
SCN8A编码的Nav1.6蛋白,在出生后发育期间逐渐取代原本Nav1.2 的定位。Nav1.6 是成年神经元远端AIS 的主要钠通道[42],并且AIS 处Nav1.6 的定位强度比体细胞和近端树突高40倍[43]。Nav1.6的超极化电压依赖性有助于AIS远端动作电位的起始;在Nav1.6 缺失的情况下,动作电位启动的阈值升高[44]。SCN8A 基因突变主要定位于Nav1.6 的跨膜段、失活门和C端,可引起Nav1.6在神经元中表达上升或下调,造成的电生理后果包括通道过早打开、通道失活受损和复苏电流升高,从而引起神经元兴奋性发生改变,导致癫痫的发生[45]。SCN8A突变已在300 多例患者中发现[46],主要以发育性癫痫性脑病(developmental and epileptic encephalopathies,DEE)为主。SCN8A-DEE 约占全部癫痫性脑病的1%,临床特征为早发型TRE、严重智力障碍、运动障碍以及较高的病死率,大部分SCN8A-DEE患者预后不佳,因此未来的研究重点是开发新型靶向治疗药物[47]。
2.2.2 VGSC的数量、结构以及功能改变导致TRE
耐药性的形成可能与AED 作用的Nav1.1、Nav1.2 或Nav1.6 等发生数量或结构性改变有关,如癫痫反复持续发作可能会导致离子通道mRNA 水平出现上升或下降,改变离子通道的密度或其中某种亚单位的组成[48]。
关于离子通道数量或结构改变的研究不多,近年来的研究更侧重于离子通道的功能改变,其中作为药物靶点的VGSC 功能发生改变,是产生耐药性的重要原因之一,尤其是偶联蛋白对Na+电流的调节。临床上广泛使用的AED 苯妥英钠和卡马西平在治疗浓度下对VGSC有抑制作用,所导致的Na+电流衰减被认为是其治疗产生效果的主要机制[49]。Nav1.1 和Nav1.2 引发的非激活、持久性的钠电流,受G 蛋白β、γ亚基调节而增加,进一步调控神经元的整合功能[50]。癫痫发作可能触发第二信使级联诱导离子通道改变[51]或胞内蛋白的磷酸化导致其对AED的反应效能减弱。同时,与钠离子通道存在相互作用的膜内结合蛋白也可能影响到VGSC 对AED敏感性的变化,与TRE的形成有关。Nav1.6与锚定结合序列AnkG存在相互作用,AnkG的膜结合结构域可增强Nav1.6 的持续钠电流[52];PRRT2 是Nav1.2 和Nav1.6 通道的重要负调控因子,与Nav1.2和Nav1.6存在特异性的相互作用,可以调节离子通道的膜表达,显著降低Na+电流[53]。此外,还有研究表明癫痫持续状态会引起钙离子的快速内流,导致线粒体除极化,产生氧化自由基,导致短时间或长时间的代谢改变,影响药物靶点VGSC的敏感性[54]。
3 展望
在过去30年里,超过15种第3代AED的问世为医生和患者提供了更多治疗多种癫痫的选择。尽管70%~80%的新发癫痫患者使用当前的AED 进入缓解期,但这些药物依然无法控制其他20%~30%患者的癫痫发作[55]。此外,尚无证据表明AED可在患者首次癫痫发作前预防癫痫的发展,这些药物似乎仅是抑制癫痫发作。AED 也不能逆转TRE 的发展,不能治疗合并症或在整体意义上减轻疾病负担。然而经过长久的研究,目前对癫痫的发生机制和耐药原因均有了深入的理解,这为发现和开发更有效的AED提供了基础。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
世界科学技术-中医药现代化(2022年2期)2022-05-25 13:17:14
中老年保健(2021年3期)2021-12-03 02:32:25
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:36
中国生殖健康(2020年2期)2021-01-18 02:51:26
中国生殖健康(2020年7期)2020-12-10 07:48:51
小学生导刊(2018年13期)2018-06-29 03:49:00
医学研究杂志(2015年7期)2015-06-22 11:01:36
同位素(2014年2期)2014-04-16 04:57:16
河南医学研究(2014年5期)2014-02-27 14:52:41
