
TEMPI综合征1例
2023-05-22 02:27苏池肖兵
疑难病杂志 2023年5期
苏池,肖兵
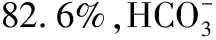
讨 论TEMPI综合征是一种新命名的罕见浆细胞疾病,2011年才被首次描述,其临床表现为以下五联征:(1)毛细血管扩张(telangiectasis,T);(2)EPO浓度升高和红细胞增多(elevated EPO and erythrocytosis,E);(3)单克隆丙种球蛋白血症(monoclonal gammopathy,M);(4)肾周积液(perinephric fluid collections,P);(5)肺内分流(intrapulmonary shunting,I)[1-4]。本例患者的临床特点包括皮肤改变、红细胞增多、M蛋白血症、多浆膜腔积液、EPO显著升高,同时多发性骨髓瘤、POEMS综合征诊断不能确立。本例患者有显著的毛细血管扩张体征,血清中EPO浓度高于检测上限,并有显著红细胞增多症且无法用其他疾病解释,具有IgG κ型M蛋白血症,CT提示肾周大量积液,均符合TEMPI综合征诊断。因患者未行静脉血气分析,无法准确计算肺内分流量,但根据患者显著呼吸困难的临床表现及动脉血气提示Ⅰ型呼吸衰竭,且无法用其他病因来解释,可以推测患者存在肺内分流。根据最新提议的TEMPI综合征诊断标准,本例患者确诊为TEMPI综合征。
TEMPI综合征是新近命名的血液系统综合征,迄今全球仅数十例报道。大部分患者首发症状为红细胞增多与毛细血管扩张,极易误诊为真性红细胞增多症(polycythemia vera,PV),临床拟诊为PV的患者需考虑TEMPI综合征可能[5]。此外,单克隆丙种球蛋白(M蛋白)血症是TEMPI综合征的特征之一,需与多发性骨髓瘤(multiple myeoma,MM)和POEMS综合征鉴别[6]。TEMPI综合征骨髓浆细胞比例通常低于10%且肾功能一般正常,M蛋白通常为IgG κ型,而POEMS综合征则通常为λ型M蛋白血症[7]。提高对TEMPI综合征的认识是及时、准确诊断的重要前提,对于红细胞增多的患者应积极完善EPO水平检测与PV相关基因突变检测,对于有M蛋白血症的患者应积极完善骨髓穿刺、VEGF水平检测与血/尿免疫固定电泳以明确M蛋白类型。
TEMPI综合征的发病机制尚不明确。起初认为肾周积液导致肾脏受压、缺氧引起EPO升高从而导致红细胞增生,但不能解释M蛋白血症的发生。后续的研究提出浆细胞可分泌巨噬细胞移动抑制因子(MIF),促进浆细胞在骨髓内黏附并调控其分化与去分化,TEMPI综合征患者骨髓、骨髓浆细胞与外周血中MIF水平显著高于健康人群。MIF可以诱导VEGF和EPO的表达,从而促进血管新生、血管扩张;并且MIF可诱导一氧化氮(NO)的产生而导致肺内分流和毛细血管扩张并促进腹腔、肾周积液。此外,研究发现TEMPI综合征患者的浆细胞具有22q11.23基因重复,可能导致MIF基因的上调。以上研究提示22q11.23基因重复导致的MIF表达上调可能是TEMPI综合征的重要发病机制[3]。
临床医师对TEMPI综合征认识严重不足,更多的病例报道有助于提升临床医师对该综合征的关注,以提升患者的诊断成功率[8]。起初TEMPI综合征的治疗通常以对症治疗为主,后续随着更多的病例总结发现M蛋白是其特征性表现,因此最新的WHO疾病分类将其归类于“浆细胞肿瘤伴副肿瘤综合征”。随后,靶向浆细胞的多种治疗方案均被应用于TEMPI综合征的治疗。文献报道硼替佐米、达雷妥尤单抗、来那度胺、高剂量马法兰及自体干细胞移植均能有效控制病情[9]。随着临床对该综合征的认识加深,未来更多的患者将得到明确诊断,前瞻性的临床试验将有助于规范治疗方案的探索。
猜你喜欢
心电与循环(2021年4期)2021-11-29
中国临床医学影像杂志(2021年5期)2021-08-13
天津医科大学学报(2021年3期)2021-07-21
重庆医学(2019年22期)2019-12-03
中学数学杂志(2019年9期)2019-05-29
中国医科大学学报(2018年7期)2018-07-06
中国眼镜科技杂志(2016年17期)2016-10-24
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
实用皮肤病学杂志(2015年4期)2015-12-22
医学理论与实践(2015年8期)2015-02-09
