
含硼药物的研究进展
2023-05-12 00:45杜丰华董正川陈乐园侯文彬李祎亮
中国药科大学学报 2023年2期
杜丰华,董正川,陈乐园,侯文彬,李祎亮*
(1天津中医药大学,天津 301617;2天津红日药业股份有限公司,天津 301700;3中国医学科学院 北京协和医学院放射医学研究所,天津市放射医学与分子核医学重点实验室,天津 300192)
近年来,在药物研发领域,经过科学家们对含硼化合物的不断探索,基于硼元素设计的药物体现出了令人期待的治疗潜力。到目前为止,已有5种含硼药物经美国食品和药品监督管理局批准上市,进入临床试验的也不在少数,同时,近年来还有新的含硼化合物被发现具有研发价值,为该领域带来了新的成果,拓宽了前景。本文将对这些含硼药物的适应证和靶点结合机制,以及代表性药物在临床试验阶段的研究进展进行概述与总结。
1 硼元素的特性
在新药研发中,研究人员对含硼化合物的研究和开发兴趣日益增加,其中的一个关键原因可能是硼元素独特的性质。硼的原子序数为5,硼原子的价电子结构式为2s22p1,它含有一个空的p 轨道,具有较强的亲电性,可以接受外来的孤对电子,进而与亲核试剂形成共价键。这一亲电特性,使得硼可与生物分子(如酶)的巯基、羟基、氨基结合并捕获它们。有机分子中的硼最常见的形式是硼酸基(R-B[OH]2),其pKa通常在7~9 之间。在生理pH 下,硼酸不带电荷,以平面三角形的sp2杂化形式存在,当环境pH 高于其pKa时,环境中水的羟基与空的p 轨道配位,形成一个配位键,硼以四面体形的sp3杂化形式存在[1-2](图1)。这一路易斯酸特性使得硼化合物可以很容易地在平面三角sp2和四面体sp3杂化状态之间相互转换,从而在与靶标作用期间采用各种结合模式,形成三方共价、四方共价或双齿共价加合物,得益于这一性质的硼在共价药物设计方面发挥了重要作用。
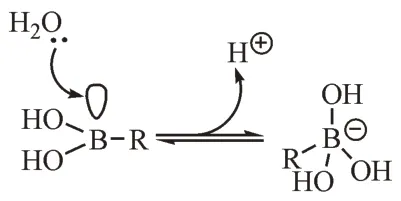
图1 硼酸从sp2到sp3杂化类型的转化
2 批准上市的含硼药物
随着首个含硼药物硼替佐米的批准,研究人员陆续研发出新颖的含硼药物,目前全球已有5种含硼药物获批上市(见表1及图2)。

表1 已获批上市的含硼药物
图2 已上市的含硼药物
2.1 硼替佐米
2005 年,千禧(Millennium)制药公司研发的硼替佐米(bortezomib,1)获得FDA 批准,成为首个获得批准上市的含硼类药物[3]。硼替佐米是第一代蛋白酶体抑制剂,用于治疗新诊断的多发性骨髓瘤和复发/难治性多发性骨髓瘤和套细胞淋巴瘤[4]。
在硼替佐米之前,研究者们起初开发了醛基多肽类蛋白酶体抑制剂,然而由于其选择性不高,生物利用度较低等缺陷,研究者们对其进行药效团替换,当原化合物中的醛基这一亲电性基团由硼酸基团替换后(图3),化合物的抑制活性显著提高,强于先导物约100 倍[5];并且,由于硼与硫之间形成的配位键不稳定,硼酸不易与半胱氨酸蛋白酶如组织蛋白酶B的巯基发生结合,这也使引入硼酸的化合物对蛋白酶体具有了独特的选择性,最终,硼替佐米从这些化合物中脱颖而出,经临床前和临床研究,最终成功上市。
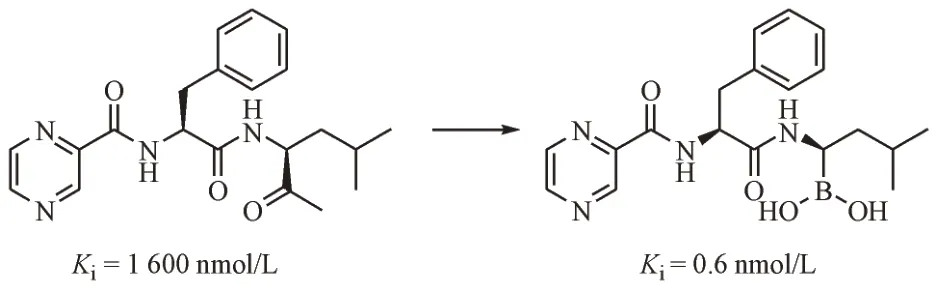
图3 硼酸基替换醛基后的蛋白酶抑制剂的活性变化
关于硼替佐米的作用机制,最主要的是泛素-蛋白酶体途径[4]。蛋白酶体存在于所有真核细胞的细胞核和胞浆中,并参与泛素化蛋白质的降解。26S 蛋白酶体是一个多亚单位酶复合体,由核心催化成分20S 蛋白酶体和两端的调控成分19S 亚基组成,20S 蛋白酶体含有3 个具有催化活性的β 亚基,即①β1、②β2、③β5 亚基,它们可对各自的蛋白底物进行水解[6]。β5 亚基(即糜蛋白样亚基)中的N-末端苏氨酸残基在水解过程中起亲核作用[7]。硼替佐米是一种二肽硼酸衍生物,该药的硼酸基团确保了对蛋白酶体的高特异性,早期研究中,该药对20S 蛋白酶体的抑制常数(Ki)为0.6 nmol/L[8]。晶体学结构(图4)显示结构中的硼原子与附近残基Thr1 的羟基形成可逆的共价结合,而硼酸基团的两个羟基分别与Thr1 和Gly47 的氨基形成氢键,进一步稳定了这一区域的蛋白质-配体复合物,同时骨架中的N,O 原子与多个残基的羰基,氨基形成氢键作用,这些作用会抑制蛋白酶体,进而对调节细胞增殖所必需的蛋白质降解造成抑制;此外,这种抑制作用还会使调节骨髓瘤细胞和骨髓基质细胞结合的信号分子失调,使得癌细胞中的促凋亡蛋白不会被降解,进而导致细胞周期停滞,引起凋亡[4]。
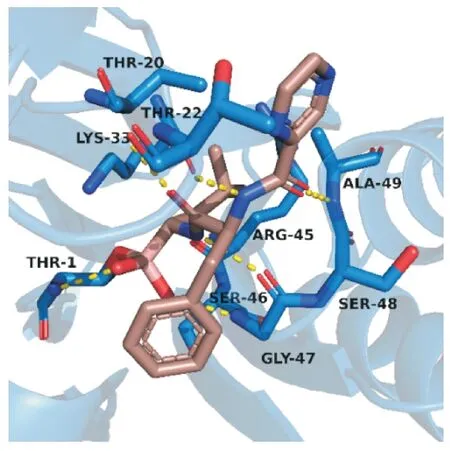
图4 硼替佐米(棕色)与20S 蛋白酶体(蓝色)的结合模式(PDB:5LF3)
在临床前的体外细胞试验中,研究者们证明硼替佐米以剂量依赖的方式对患者的多发性骨髓瘤细胞产生抑制,IC50最低为2.5 nmol/L[9]。之后,研究者们在大量的Ⅰ ~ Ⅲ期临床试验中对硼替佐米的抗肿瘤活性进行了比较系统的研究。并且,从这些临床研究中发现,硼替佐米与常规治疗剂或放射线联合使用时,具有化学/放射增敏活性,可作为克服耐药性的手段[4]。
2.2 伊沙佐米
伊沙佐米(ixazomib,2)是第二代蛋白酶体抑制剂,由千禧制药公司公司开发,2015 年首次获得批准,可与来那度胺和地塞米松联用于先前至少接受过一种治疗的多发性骨髓瘤患者[10]。
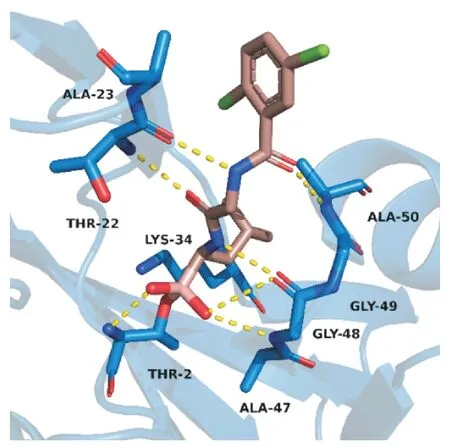
图5 伊沙佐米(棕色)与20S 蛋白酶体(蓝色)的结合模式(PDB:5LF7)
给药方式上,伊沙佐米是首个口服蛋白酶体抑制剂,每周用药1 次,减轻了患者依从性[10]。结构上,与硼替佐米类似,伊沙佐米的生物活性形式是二肽亮氨酸硼酸,在生理条件下,它由含柠檬酸酯的前药经水解生成。作用机制上,伊沙佐米优先结合20S蛋白酶体的③蛋白水解(β5)位点,通过共晶结构(图5)可以观察到,伊沙佐米的硼原子与苏氨酸残基(THR-2)发生结合,对其产生可逆性的抑制,主链中的N、O 与附近的残基形成氢键作用。在体外实验中,伊沙佐米对β5 蛋白的IC50为3.4 nmol/L,并且在较高浓度下,该药还能抑制①蛋白水解(β1)位点和②蛋白水解(β2)位点,IC50分别为31 和3 500 nmol/L[11]。同时,伊沙佐米通过增加多发性骨髓瘤细胞的凋亡来减少肿瘤进展,并破坏多发性骨髓瘤细胞与骨髓微环境的相互作用,从而减少血管生成和溶骨性病变[12]。在临床前试验中,研究者们发现伊沙佐米在多发性骨髓瘤异种移植模型中显示出抗肿瘤活性[13]。
目前,伊沙佐米正在进行进一步的Ⅲ期临床试验,其中包括用于治疗多发性骨髓瘤,以及用于治疗其他血液系统恶性肿瘤(如淋巴瘤和淀粉样变性)以及实体瘤的临床试验。在这些针对复发/难治患者、新诊断的患者和维持疗法的Ⅲ期试验中,伊沙佐米正在作为单一药物或与其他疗法联合进行评估[12]。
2.3 他伐硼罗
他伐硼罗(tavaborole,3)是安纳考尔(Anacor)制药公司开发的一种新型抗真菌药物,该药于2014年获得FDA 批准上市[14]。他伐硼罗是一种真菌蛋白质合成抑制剂,用于局部治疗因红色毛癣菌感染引起的脚趾甲真菌病[15]。真菌的亮氨酰tRNA合成酶(LeuRS)是tRNA 校对和蛋白质合成所必需的一种酶,在翻译过程中,LeuRS 负责催化亮氨酸与亮氨酸特异性tRNA(tRNALeu)的结合。他伐硼罗的苯并硼唑结构中含有硼原子,在LeuRS的编辑位点上,硼原子与tRNALeu 上的3′-末端腺苷上的2′,3′-羟基结合,形成一种稳定的tRNALeu-Tavaborole加合物(图6),从而阻断亮氨酸的催化周转,引起下游蛋白质的合成抑制,阻止了真菌的进一步生长,该药对LeuRS 的IC50为2.1 μmol/L[16]。他伐硼罗的结构是通过研究类似的硼酸酯构效关系时发现的,之后,在经过不同类型的真菌试验后,研究人员发现它具有广谱的抗真菌活性[17-18]。通过用碳取代硼后,研究人员测试其抑制真菌活性,发现降低了50 倍,证明了他伐硼罗是一种高度特异的LeuRS 抑制剂,而且苯并硼唑环上的硼原子是他伐硼罗独特的抑制机制所必需的[19]。
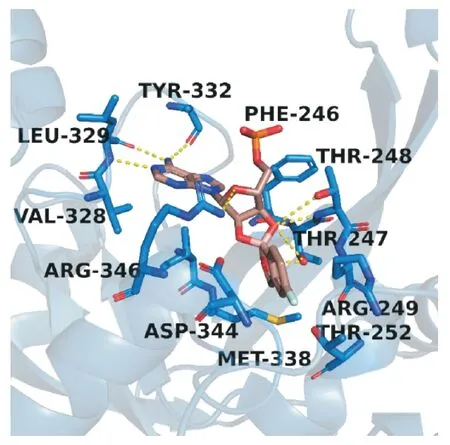
图6 他伐硼罗(棕色)与亮氨酸特异性tRNA(蓝色)的结合模式(PDB:2V0C)
在临床研究中,他伐硼罗显示出良好的指甲穿透性质和疗效。在Ⅰ/Ⅱ期临床试验中,15 名甲真菌病患者接受了7.5%的他伐硼罗治疗,结果表明,他伐硼罗能有效穿透指甲,治疗水平的他伐硼罗在最后一次给药后效果至少持续3个月。另外,在3 个独立的Ⅱ期剂量范围研究中,5%的给药浓度被认为是进一步研究的最佳浓度[15]。随后进行的Ⅲ期临床试验评估了5%他伐硼罗与赋形剂相比的疗效和安全性。患者的真菌学结果显示,5%的他伐硼罗溶液被证明比赋形剂更有效,涂抹部位反应的发生率很低,证明其安全性较好[20]。
2.4 克立硼罗
克立硼罗(crisaborole,4)由安纳考尔制药开发,是一种非甾体类选择性PDE4 抑制剂,药用剂型为2%的外用软膏,该药于2016 年获得FDA 批准,用于治疗轻度至中度特应性皮炎[21]。
环磷酸腺苷(cAMP)相关的信号级联在调节免疫功能中起着关键的作用[22],磷酸二酯酶4(PDE4)特异性催化cAMP 的水解,是免疫和炎症细胞中主要的磷酸二酯酶[23],其水平在特应性皮炎患者中会升高[24]。克立硼罗结构中硼的存在使其在PDE4的催化位点模拟了cAMP中的磷酸盐结构,以四面体的形式对PDE4 的催化活性部位产生竞争性抑制[25],该抑制将引起细胞内cAMP 的增多,进而激活蛋白激酶A 和其他下游信号通路,如细胞核因子κB(NF-κB)通路及活化T 细胞核因子(NFAT)通路,从而抑制炎症细胞因子(如TNF-α和各种白介素)的产生,起到治疗特应性皮炎的效果[26]。在早期研究中,克立硼罗对PDE4 的IC50为0.49 μmol/L[27]。
此外,独特的硼唑结构使得克立硼罗具备合理的理化性质,更易穿透表皮和真皮到达炎症部位。并且,克立硼罗独特的局部配方允许其在炎症和快速新陈代谢部位进行靶向抑制,之后可以快速代谢成无活性化合物进行排泄,从而避免全身暴露和潜在的非目标副作用[28]。
4 项Ⅰ期和Ⅱ期研究分析了儿童和成人使用克立硼罗的药代动力学、疗效和安全性,结果显示,2%的克立硼罗外用软膏显示出良好的耐受性,包括在最大剂量条件下,表现出有限的全身性暴露[29-32]。根据一项Ⅲ期试验的结果,长期连续使用克立硼罗后,相当大比例的轻中度特应性皮炎患者的病情得到改善。并且,在延长使用克立硼罗的周期内,患者耐受性良好,未观察到安全信号,这表明患者需要更长的治疗期才能控制病情,并安全使用克立硼罗[33]。
2.5 美罗培南-法硼巴坦
美罗培南-法硼巴坦(meropenem-vaborbactam)是2017 年FDA 批准的组合药物,用于治疗复杂尿路感染[34]。它由β-内酰胺酶抑制剂法硼巴坦和碳青霉烯类抗生素美罗培南组成,虽然法硼巴坦本身不是抗生素,但它与美罗培南联合使用可以防止其被β-内酰胺酶水解[35]。法巴硼坦(vaborbactam,5)由Rempex 制药公司研发,是一种基于环状硼酸药效团的新型β-内酰胺酶抑制剂,其结构中含有的硼可与碳青霉烯酶(KPC)中的酶活性位点形成配位键。从机制上讲,硼与催化丝氨酸残基(SER-45)上的羟基形成四面体加合物,模拟了酶水解时的过渡态,对活性位点具有高亲和力,产生竞争性抑制[36]。结晶学研究(图7)证实了它与催化丝氨酸的共价结合,而结构中的羧基在底物口袋中形成多组氢键,酰胺羰基与附近残基的两个氢键供体基团形成氢键[37]。这些作用力使得它对于KPC 具有极佳的抑制活性。此外,法硼巴坦不受KPC 变异、KPC 酶突变或外排机制的影响[38]。美罗培南具有广谱的体外活性、良好的安全性和治疗严重革兰氏阴性菌感染的效果,因此被选为理想的碳青霉烯类抗生素[39]。

图7 法硼巴坦(棕色)与KPC(蓝色)的结合模式(PDB:4XUZ)(KPC:Klebsiella Pneumoniae Carbapenem,克雷伯菌中产生的碳青霉烯酶)
在临床前实验中,这种组合药物对耐药的革兰氏阴性菌,特别是产生碳青霉烯酶的肺炎克雷伯氏菌显示出较强的抗菌活性(MIC50≤ 0.06 mg/L)[40]。在药代动力学试验中,法硼巴坦与大多数β-内酰胺类抗生素曲线相似,其典型特征是半衰期短和分配量低。研究人员通过临床前试验和Ⅰ期相关试验优化了给药方案,使其在表现出杀菌活性的同时,防止了MICs 高达8 mg/L 的病原体产生耐药性,实现了PK/PD 目标[37,40]。一项Ⅲ期试验(NCT02168946)显示,与哌拉西林-他唑巴坦相比,使用美罗培南-法硼巴坦后,包括急性肾盂肾炎(AP),以及严重的耐碳青霉烯类抗药性肠杆菌(CRE)感染的复杂尿路感染患者的症状完全缓解或得到了极大的改善[41]。
3 临床研究中的含硼药物
3.1 度格列汀
度格列汀(dutogliptin,6)是由菲诺米克斯(Phenomix)公司、森林实验室公司(Forest Laboratory)和意大利凯西制药公司(Chiesi FarmPharmtici spa)联合开发的一种口服的选择性二肽基肽酶-4(dipeptidyl peptidase-IV,DPP-IV)抑制剂,对DPP-Ⅳ的抑制活性为IC50= 25 nmol/L[42],用于治疗2 型糖尿病(T2DM)[43]。二肽基肽酶-4 是一种丝氨酸蛋白酶,当此酶受到抑制后,将导致内源性葡萄糖依赖型胰岛素样多肽(GIP)和胰高血糖素样肽-1(GLP-1)活性增强,最终导致胰岛素分泌增强,从而降低血糖水平、糖化血红蛋白(HbA1c),以及胰高血糖素分泌和肝脏葡萄糖生成的减少[44]。
在一项多中心、随机、双盲、安慰剂对照的研究中,研究者对度格列汀的药代动力学和药效进行了实验和评价,接受格列酮或二甲双胍背景疗法的2 型糖尿病患者,每天口服一次度格列汀,在3 种不同剂量(100、200 或400 mg)水平下,持续4 周后,患者均表现出良好的耐受性,并且血糖控制得到了明显改善[45]。目前,度格列汀与干细胞动员剂非格斯汀(filgrastim)联合给药治疗心肌梗死的Ⅱ期试验正在进行中(NCT03486080)[46]。
3.2 阿考硼罗
被忽视疾病药物研发组织(DNDi)开发的阿考硼罗(acoziborole,7)是一种抗寄生虫药物,用于治疗人类非洲锥虫病(HAT)[47],其对布氏锥虫菌株的MIC 为0.6 μg/mL[48]。不过,目前该药的靶点和作用机制尚未得知[49]。在临床前试验中,阿考硼罗以25 mg/kg 口服给药,7 d 后对中枢神经系统的布氏锥虫感染治愈率达到了100%,表现出了显著的抗锥虫活性,以及单次口服给药治愈HAT 的可能性。同时,体内研究表明,该化合物具有高度的跨物种生物利用度,并且可以跨越血脑屏障,在啮齿类动物的脑和脑脊液中达到与治疗相关的浓度[48]。目前,阿考硼罗正处于一项研究其疗效和安全性的Ⅲ期试验中(NCT05256017)[50]。
3.3 GSK3036656
葛兰素史克(GlaxoSmithKline)公司研发的GSK3036656(8)[51]是一种作用于结核分枝杆菌的亮氨酰tRNA 合成酶(MtbLeuRS)抑制剂,用于治疗结核病[52]。与他伐硼罗的抗菌机制类似,GSK3036656 结构中的硼与tRNA3′末端核苷酸的Ade76形成两个配位键,由此产生的共价加合物将亮氨酰tRNA 合成酶的3′端束缚在非生产复合物的编辑位点上,进而阻止亮氨酸化,从而抑制了细菌蛋白的合成。位于C-3 侧链的氨基可以形成3个氢键,在结合中起重要作用。此外,Cl 原子的存在显著改善了它的抑制活性和抗结核作用,并对其他细菌产生了选择性。该药有很强的酶抑制活性(IC50= 0.2 μmol/L)与体外抗结核活性(MIC =0.08 μmol/L)。同时,在小鼠结核病感染模型中,该药具有显著的口服生物利用度和低剂量体内疗效[51]。在一项首次在人体内(FTIH)进行的研究(NCT03075410)中GSK3036656 在单次和重复剂量的口服给药后表现出良好的安全性及耐受性[53]。目前,一项关于该药的Ⅱ期试验正处于尚未招募的阶段(NCT05382312)[54]。
3.4 AN2898
与已经批准的克立硼罗类似,辉瑞(Pfizer)公司开发的AN2898(9)是另一种用于治疗特应性皮炎的磷酸二酯酶4(PDE4)抑制剂,具有竞争性抑制、可逆性结合的特点;通过抑制肿瘤坏死因子α(TNF-α)、白细胞介素IL-12、IL-23 等相关细胞因子的释放达到治疗特应性皮炎和银屑病的效果[55]。从图中可以看出,AN2898 相比于克立硼罗多了一个氰基,这一邻位氰基的引入也使得AN2898 对PDE4 的抑制活性得到了提高(IC50= 60 nmol/L)[56]。
关于AN2898 的临床试验正在进行中,在一项多中心、随机、双盲、对照、双侧对照的Ⅱ期研究中(NCT01301508),特应性皮炎患者接受AN2898 软膏(1%)治疗,持续6 周,试验终点表明,AN2898 软膏(1%)在降低特应性皮炎病变的严重程度和症状方面具有较好的效果,且安全性和耐受性良好[57]。
3.5 他尼硼巴坦
他尼硼巴坦(taniborbactam,10)是一种含硼的全谱β-内酰胺酶抑制剂,具有可逆性、选择性抑制的特点。维纳托克斯(venatorx)制药公司的研发人员经筛选得到含硼先导化合物,基于结构进行药物设计,并对构效关系研究后,发现了该化合物,其结构可通过与羟基和锌离子结合模拟酶水解时的过渡态,对丝氨酸类β-内酰胺酶(SBLs)和金属类β-内酰胺酶(MBLs)产生抑制。从晶体结构中可以观察到[58],环状硼酸酯中的硼、氧原子和羧基中的氧原子与金属类β-丝氨酸酶NDM-1活性中心的两个锌离子发生结合(图8-A),而在与OXA-10 的晶体模式(图8-B)中,硼原子共价连接到具有亲核性的残基Ser67 上。在临床前试验中,他尼硼巴坦对所有主要的临床相关的β-内酰胺酶表现出亚微摩尔级的半数最大抑菌浓度(表2)[59]。他尼硼巴坦是首个进入临床试验的含硼全谱β-内酰胺酶抑制剂。一项随机、双盲、安慰剂对照的Ⅰ期试验(NCT02955459)中,研究者测试了在不同剂量下他尼硼巴坦在健康受试者中的安全性和药代动力学特性,结果显示,该药表现出线性和剂量比例的PK曲线,变异性低,安全性良好[60]。
目前,一项评估他尼硼巴坦联合头孢吡肟治疗尿路感染的疗效、安全性和耐受性的Ⅲ期临床试验按计划已于2021年底完成(NCT03840148)[61]。
3.6 GSK8175
GSK8175(11)属于第二代针对丙型肝炎病毒(HCV)的RNA 聚合酶抑制剂,主要针对丙型肝炎病毒(HCV)的RNA 聚合酶发挥抗病毒作用。葛兰素史克公司的研发人员对一种血浆半衰期较短的临床候选药GSK5852 的代谢性质进行优化,最终发现了GSK8175。X 射线衍射数据显示其结构中的关键药效团N-苯基硼酸在高度有序的水分子介导产生的氢键相互作用下,与HCV 的NS5B 蛋白(如GT1a 316Y蛋白)发生结合,对基因型1a(GT1a)的复制子可以产生有效抑制(EC50= 32 nmol/L)。临床前的研究显示经过几轮结构修饰后,体外和细胞分析证实了GSK8175 的有效活性,体内研究显示其在大鼠体内的低清除率证实了其优越的药代动力学特征[62]。目前,研究人员在一项第Ⅱ阶段(NCT02014571)的临床试验中,已评估了在20 mg口服剂量下,GSK8175 治疗慢性丙型肝炎病毒的安全性和有效性[63]。
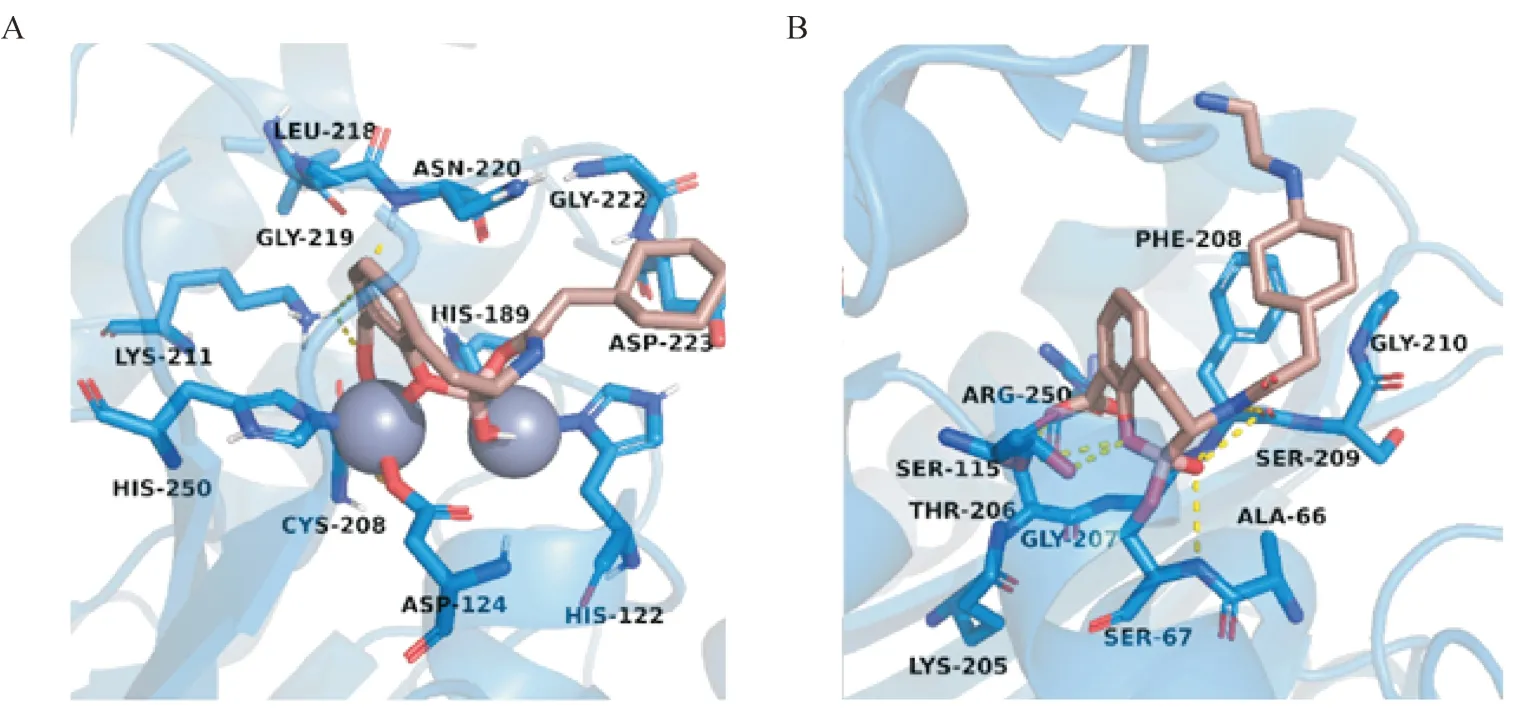
图8 A:他尼硼巴坦(棕色)与金属β-内酰胺酶NDM-1(蓝色)的结合模式(PDB:6RMF);B:他尼硼巴坦与丝氨酸β-内酰胺酶OXA-10的结合模式(PDB:6RTN)
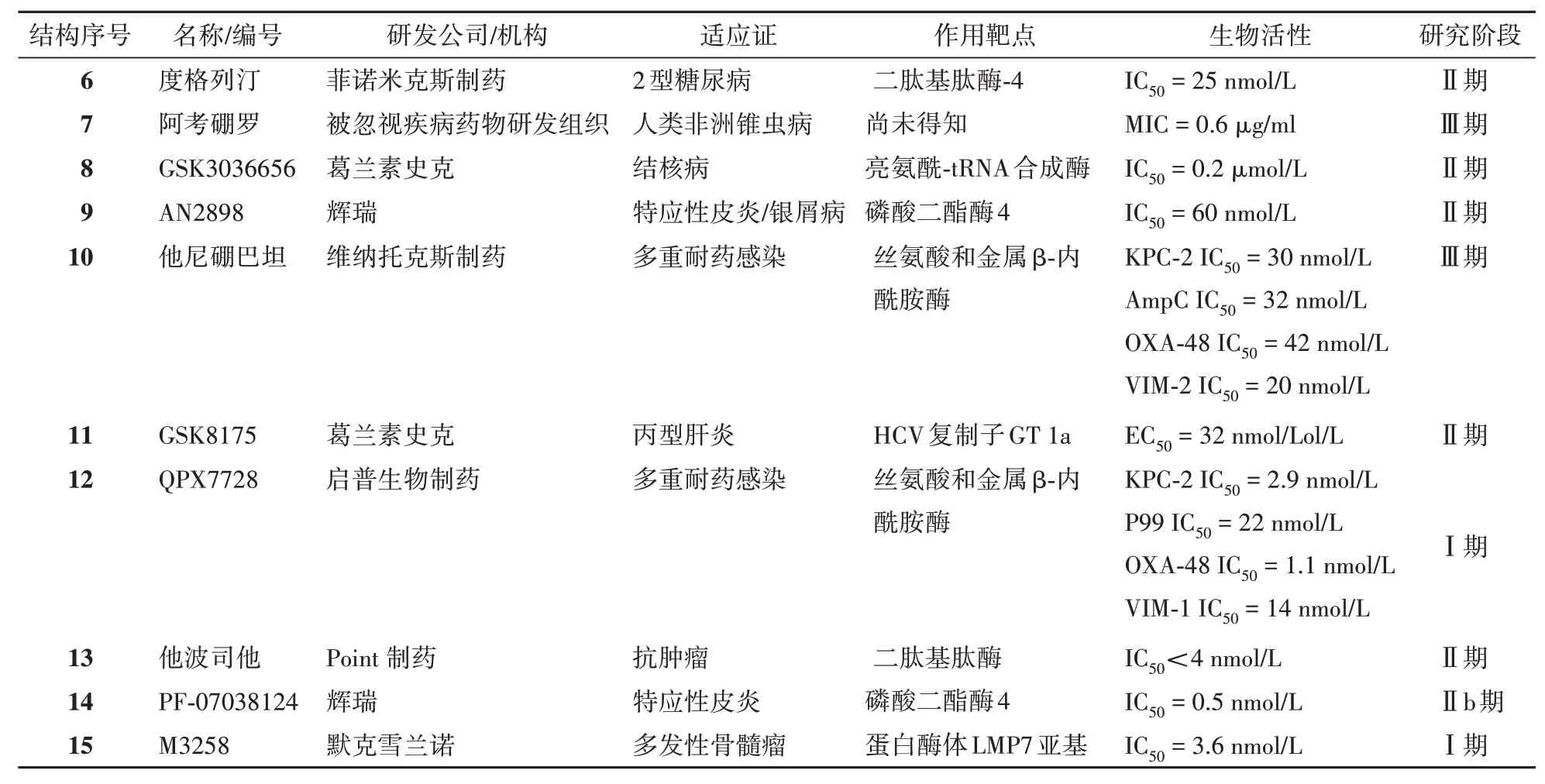
表2 临床研究中的含硼药物
3.7 QPX7728
启普生物制药(Qpex Biopharma)公司开发的QPX7728(12)是一种超广谱β-内酰胺酶抑制剂,可以与β-内酰胺类抗生素结合使用来治疗耐多药的革兰氏阴性细菌感染。该药由研发人员通过对含双环硼酸的先导化合物进行修饰后,逐步获得抗菌谱后最终得到。QPX7728 对绝大多数β-内酰胺酶表现出广泛的抑制活性(表2),不仅包括A 类和C类丝氨酸酶,D类苯唑西林酶(OXA酶),还包括B类金属酶,如VIM-1,并且几乎不受孔蛋白修饰和外排的影响[64]。在体内活性研究中,QPX7728表现出与β-内酰胺类抗生素相似的药代动力学性质,并且不需要成前药就能达到良好的口服生物利用度[65]。
在一项多药联用试验中,QPX7728 与美罗培南、头孢吡肟或头孢噻嗪联用后,对临床分离的铜绿假单胞菌产生了高度有效的抑制活性[66]。一项随机、双盲、安慰剂对照的Ⅰ期临床试验(NCT04380207)已于2022 年完成,该试验对单剂量和多剂量递增使用的QPX7728 的安全性、耐受性和药代动力学进行了研究[67]。
除了以上代表性的7种,近几年还有几种含硼药物进入了临床试验阶段,包括针对DPP-IV 靶点的他波司他(13,IC50< 4 nmol/L)[68-69],以PDE-4 为靶点的PF-07038124(14,IC50= 0.5 nmol/L)[70]和靶向抑制免疫蛋白酶体LMP7 的M3258(15,IC50=3.6 nmol/L)[71](见表2及图9)。
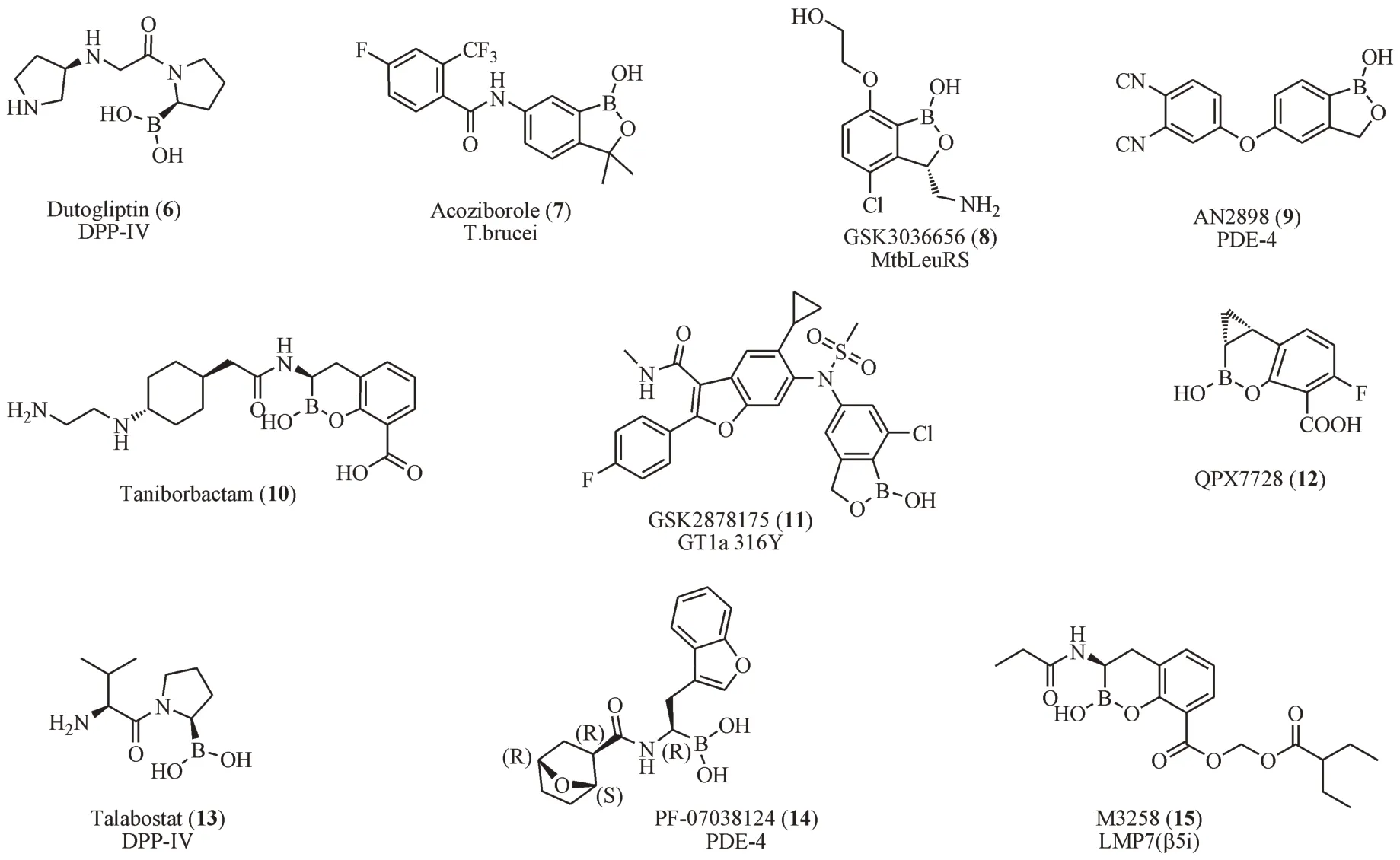
图9 进入临床试验阶段的含硼药物
4 近年新发现的含硼药物
除了已上市和临床研究中的含硼药物之外,近年来不断有新的含硼化合物在相关研究中被发现,它们目前尚处于临床前阶段或生物活性评价阶段其中包括国内的科研人员在药物研发方面取得的令人惊喜的成果。
Zhang 等[72]研究了一类7-丙酰胺苯并硼唑化合物的合成和构效关系,发现它们其中的两种化合物(16,17)对卵巢癌具有良好的抑制活性。Wang 等[73]应用药效团融合策略,基于[1-(3′-硫代丙氨基)甲基]硼酸设计了一种新型结构(18),它对代表性的金属和丝氨酸-β 内酰胺酶(MBL/SBL酶)表现出广谱的抑制作用,这将有助于开发针对临床上MBL/SBL 酶的新型双重抑制剂。Ju 等[74]发现了一种新型硼酸修饰的酪蛋白水解酶P(ClpP)抑制剂(19),它能够靶向结合金黄色葡萄球菌细胞中的内源性ClpP(SaClpP),对其有明显的抑制作用。安纳考尔制药的研究者们[75]发现了一种新型的酰胺类吡嗪氧基苯并硼唑类化合物(20),它表现出了良好的抗疟活性和药代动力学特性,且无遗传毒性,适合临床前开发。
Tan 等[76]利用从头文库设计和虚拟筛选技术,发现了一类含α-氨基硼酸的人酪蛋白水解酶P(hClpP)抑制剂(21,22,23),它可以使错误折叠蛋白质发生降解,其IC50分别在(10.2 ± 3.7)μmol/L,(8.4 ± 2.6)μmol/L 和(12.4 ± 0.1)μmol/L 的低微摩尔范围内,而且这3 种化合物在hClpP 存在的情况下,对人酪蛋白水解酶XP(ClpXP)的IC50分别为(0.8 ± 0.3)μmol/L,(0.9 ± 0.4) μmol/L 和(1.0 ±0.3)μmol/L,这使其成为首类被报道的hClpXP 抑制剂。Clark等[77]通过针对自体趋化蛋白(ATX)的化合物进行构效关系的分析,设计合成了一种基于内源性类固醇调节剂的V 型ATX 抑制剂(24),带有含硼弹头的该类结构以疏水空腔和磷酸二酯酶结构域的活性部位为靶点,竞争性抑制该酶活性,调节溶血磷脂酶(LPA)表达,对ATX-LPA 信号通路产生影响。Mowbray 等[78]开发了一种含三氮唑与酰胺结构的苯并硼唑化合物(25),该化合物具有强大的体外抗利什曼原虫活性和良好的代谢稳定性。它主要通过抑制利什曼原虫的切割和多聚腺苷酸化特异性因子(CPSF3)核酸内切酶发挥作用,目前该化合物已被提名为临床前候选药物(DNDI-6148)。近年新发现的含硼化合物结构及靶点等见表3及图10。
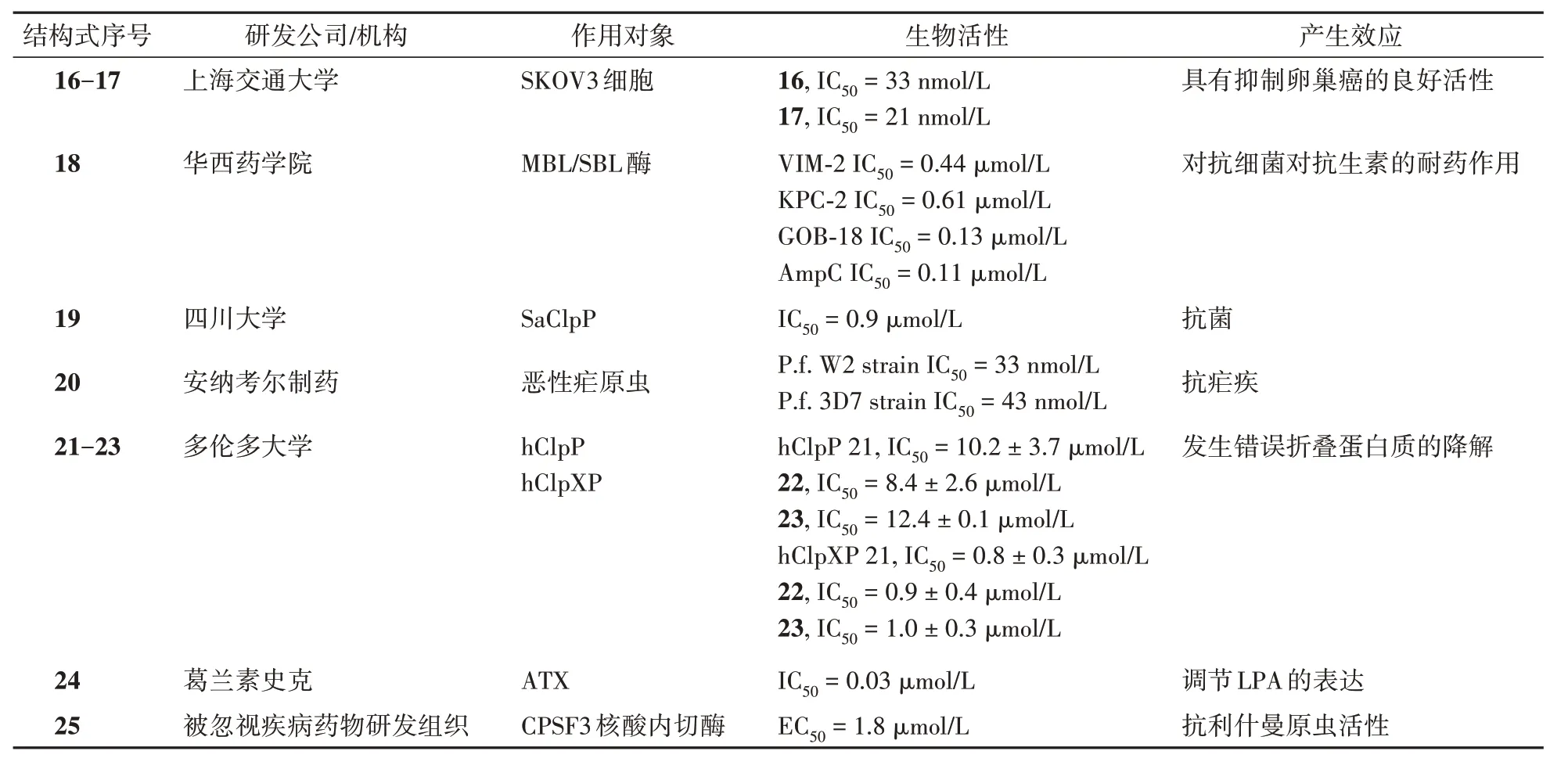
表3 近年新发现的含硼药物

图10 近几年新发现的含硼药物
5 总结与展望
硼原子在化合物结构中的出现增加了药物分子的多样性。硼原子含有一个空轨道,具有路易斯酸性,可与路易斯碱形成络合物,同时,硼表现出的亲电性使其可与亲核基团包括酶残基中常见的羟基,氨基等氨基酸基团形成共价键。因此,药物在引入硼原子后可通过形成可逆的共价键与生物靶标结合,这一作用比多数药物典型的非共价、疏水相互作用具有更大的稳定性,提供了额外的亲和力,一定程度上增强了药物对靶标的生物活性。但结构中含有硼酸的药物稳定性较差,可能会在与靶标结合之前被降解。另外,含硼药物进入体内以后,也可能表现出与其他内源性亲核靶标的非特异性反应,如与丝氨酸、赖氨酸等氨基酸残基形成较强的作用力,产生脱靶效应,导致药物的不良反应。因此,这需要研发者在开发过程中对硼进行结构形式上的优化,如改造为硼酸酯和含硼杂环等;以及开发出使药物特异性靶向的策略等。
在硼替佐米获得批准上市之后,新的含硼化合物被不断设计并用于药物开发中,为药物研发提供了新途径。针对不同靶点,具有不同适应证的含硼药物不断被研发人员发现,并以良好的活性和成药性被应用到进一步的临床研究中。当然,该类药物被发现具有活性的同时,其作用方式和特性等还需继续探索,比如一类苯并硼唑类化合物具有抗疟疾作用,但其作用靶点与机制目前仍需寻找并阐明。
含硼类药物在新药开发中具有广阔的前景,研究者们对这类药物的研究也在不断深入,我们相信,随着新化合物结构的出现以及更多靶点和作用机制的阐明,未来将会有更多安全高效的含硼药物进入临床治疗的领域中。
猜你喜欢
无机盐工业(2022年2期)2022-02-21
云南化工(2021年7期)2021-12-21
高科技纤维与应用(2021年2期)2021-04-04
实用药物与临床(2020年10期)2020-12-29
山东化工(2020年4期)2020-03-30
中国临床医学(2019年2期)2019-05-21
华东师范大学学报(自然科学版)(2018年2期)2018-05-14
科学中国人(2017年36期)2017-06-09
中国病理生理杂志(2015年8期)2015-12-21
哈尔滨医药(2015年6期)2015-12-01
