
不同FGFR1突变与先天性低促性腺激素性性腺功能减退症的相关性
2023-05-04 02:42杨宇帆伍学焱茅江峰
基础医学与临床 2023年5期
杨宇帆,王 曦,聂 敏,伍学焱,茅江峰
中国医学科学院 北京协和医学院 北京协和医院 内分泌科,北京 100730
先天性低促性腺激素性性腺功能减退症(congenital hypogonadotropic hypogonadism,CHH)是一类由多种基因突变导致的下丘脑促性腺激素释放激素(gonadotropin-releasing hormone,GnRH)分泌减少或作用缺陷引起的先天性疾病,男性发病率约1/10 000,女性更低。疾病主要表现为青春期不发育和生育障碍[1]。
目前发现成纤维细胞生长因子受体1(fibro-blast growth factor receptor 1,FGFR1)、促性腺激素释放激素受体(gonadotropin releasing hormone receptor,GnRHR)、白细胞介素17受体D(interleukin-17 receptor D,IL17RD)等30余个基因与CHH发病有关。FGFR1突变导致CHH较为常见,占全部患者8%~10%,符合常染色体显性遗传模式[2]。FGFR1位于染色体8p11.23,包含18个外显子,编码的FGFR1蛋白属于酪氨酸激酶受体超家族成员,在体内广泛表达[3]。
新近的研究发现,携带相同FGFR1突变的个体,临床表现差别很大。同一家系内的携带者可表现正常性腺轴功能,也可表现为CHH[4]。因此仅以FGFR1突变难以解释疾病发生。本研究拟通过对FGFR1突变CHH患者多个基因进行筛查,寻找是否同时存在其他基因突变,共同导致疾病发生。
1 材料与方法
1.1 病例
2013年5月至2021年5月在北京协和医院内分泌科门诊随访的CHH患者共160例。同时满足以下条件可诊断CHH。1)男性年龄≥18岁或女性年龄≥16岁仍无第二性征发育;2)男性睾酮(testosterone,T)<1 ng/mL 或女性雌二醇<25 pg/mL,且黄体生成素(luteinizing hormone,LH)和卵泡刺激素(follicle-stimulating hormone,FSH)水平均低下;3)其余垂体前叶激素水平正常;4)鞍区磁共振成像(magnetic resonance imaging, MRI)正常;如果患者同时存在嗅觉丧失,可诊断为卡尔曼综合征(Kallmann syndrome,KS)。嗅觉功能正常的CHH患者可诊断为嗅觉正常的CHH(normosmic CHH,nCHH)。经全部患者和部分患者父母知情同意,抽取外周血,提取基因组DNA进行二代测序panel分析。研究通过北京协和医院伦理委员会审批(编号JS-2111)。
1.2 方法
1.2.1 临床资料收集:包括患者的诊断、年龄、性别、睾丸体积、基础LH、FSH和T水平(北京协和医院检验科测定)。
1.2.2 基因的检测:按照操作说明书,提取患者及父母外周血白细胞DNA,使用高通量测序系统Illumina Nextseq 500对产物进行panel二代测序,对FGFR1突变进行Sanger测序验证突变来源。
1.2.3 测序结果分析:二代测序的定制化基因panel包含CHH相关的基因97个。参考基因组版本为GRCh37/hg19,并参考千人基因组、外显子组测序项目 (ESP6500)、外显子组聚集联盟(The Exome Aggregation Consortium,EXAC)和EXAC-EAS(EXAC约4 000个东亚人)的数据库。将致病基因的测序结果,与人类基因突变数据库(Human Gene Mutation Database,HGMD)进行比对。
1.2.4FGFR1突变的致病性分析:使用1 000 g 2015aug_all、GnomAD判断新发突变的频率,GERP评估突变位点的氨基酸保守性。所有突变使用美国医学遗传学与基因组学学会(The American College of Medical Genetics and Genomics, ACMG)指南[5]判断致病性。使用SIFT,PolyPhen_2,MutationTaster,REVEL,MCAP,LRT这6种生物信息学软件判断FGFR1和其他CHH相关基因错义突变的致病性。从未报道过的突变、或在NCBI、Ensembl、 Exome Variant Server和500 normal Chinese controls中没有发现的突变,或在上述数据库中等位基因频率≤0.001,同时不少于3种生物信息学软件预测为有害,可认为该突变存在致病性,认定为可能致病。
1.2.5 基因相互作用分析:使用Genemania研究FGFR1与其他CHH相关基因之间的作用。使用String检测蛋白作用。通过Gene Ontology分析寻找基因作用通路。
2 结果
2.1 FGFR1突变分布与致病性分析
9.3%(15/160)的CHH患者 (男/女=11/4;nCHH/KS=8/7)中共发现15个罕见FGFR1突变(表1)。其中10个为错义突变,包括p.V273M[6]、p.P366L[7]、p.R254W[8]、p.R424H[6]、p.L326F、p.S125L、p.M1I、p.R80C、p.G260R和p.T761I。(前4个已被报道,其余为新发突变)。5个可能导致蛋白发生截断的突变:c.536delC、c.838dupT、c.54_55del、c.250_264delGAGGAGGTGGAGGTG、c.325_342delAGTGACACCACCTACTTC(均为新发突变)。根据ACMG指南、SIFT等6种生物信息学软件和GREP软件预测结果,这些突变在排除多态性后均被判定为可能致病性。
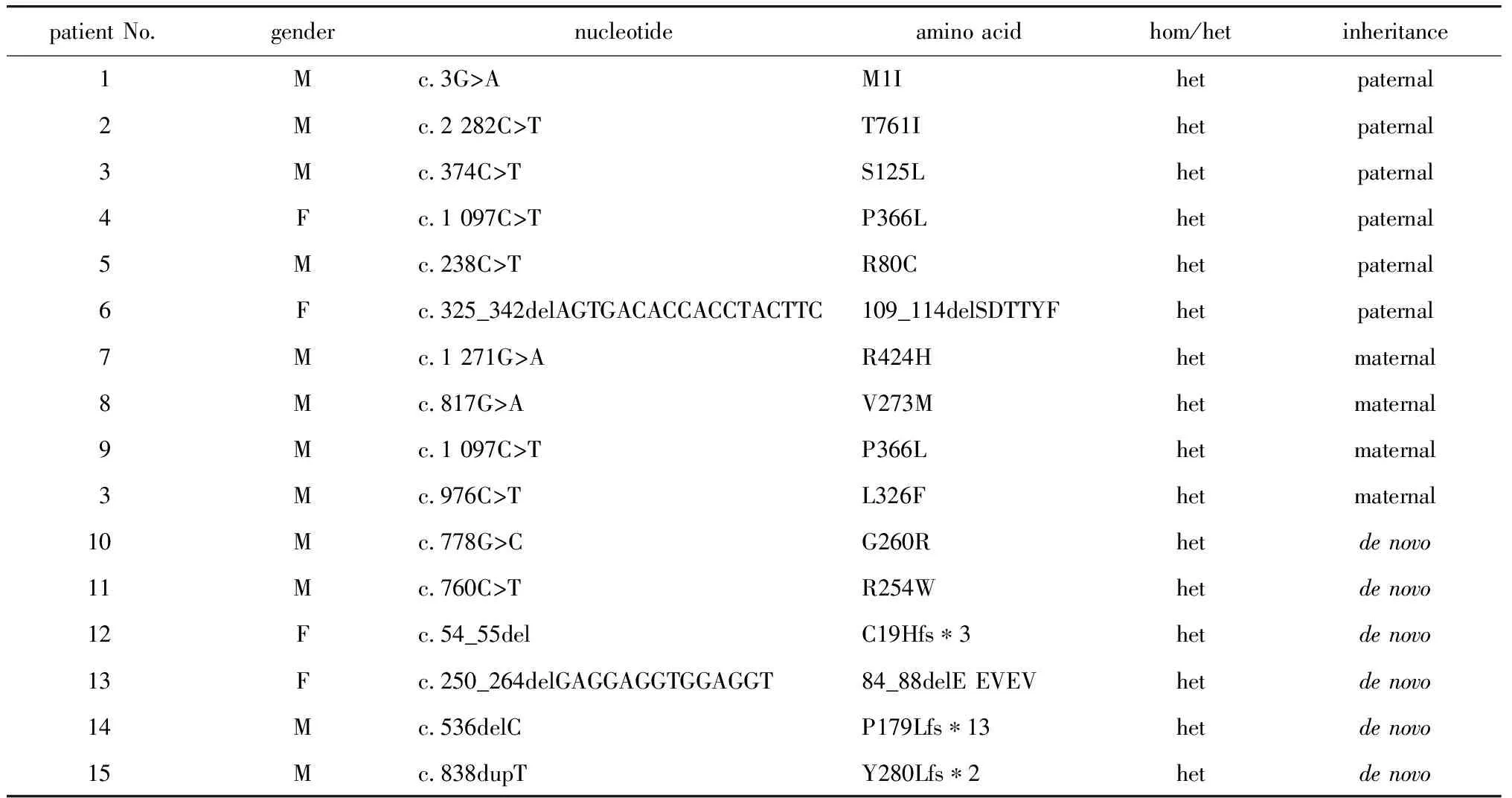
表1 FGFR1突变的来源Table 1 Inheritance of FGFR1 mutations
FGFR1突变来源分析:有9例(男性7例,女性2例)FGFR1突变来自父/母(遗传突变组)。有6人为自发突变(自发突变组)。 遗传突变组的FGFR1的突变主要为错义突变,只有1例是缺失突变(109_114delSDTTYF),而自发突变组的突变类型包括3例的截断突变(C19Hfs*3、P179Lfs*13、Y280Lfs*2)、1例缺失突变(84_88delEEVEV)及2例错义突变。
15例FGFR1突变的CHH患者父母均为正常表型。
2.2 其他CHH相关基因突变来源与致病性分析
在遗传突变组(n=9)中,6例患者还同时存在另一个基因的突变(表2)。除FGFR1突变外,其他突变位点分别为前动力蛋白受体2(prokineticin receptor 2,PROKR2) (W178S)、成纤维细胞生长因子8(fibroblast growth factor 8,FGF8)(T121N)、硫酸肝素6-O-磺基转移酶1(heparin sulfate 6-o-sulfotransferase 1,HS6ST1)(P242L)、信号素3A(semaphorin 3A,SEMA3A)(R734W)、亮氨酸拉链样转录调节因子1(leucine-zipper-like transcription regulator 1,LZTR1)(c.21delG)。根据ACMG指南,LZTR1(p.Q10rfs *15)可能导致蛋白截短而判定为致病性突变。PROKR2(W178S)已有多次致病性突变的报道[9]。SEMA3A(R734W)、FGF8(T121N)和HS6ST1(P242L),经过SIFT等6种生信软件和GREP软件分析,均判定为致病性。此外,有1例患者是FGFR1复合杂合突变。这些基因突变,来自表型正常的父亲或母亲。

表2 多基因位点突变的CHH患者Table 2 Multiple gene mutations in CHH patients
利用Genemania软件分析这些基因的相互作用FGFR1和其他基因(PROKR2、SEMA3A、FGF8和LZTR1)之间存在复杂相互作用。FGFR1与FGF8关系最为密切(图1A)。String蛋白相互作用分析表明,FGFR1蛋白与HS6ST1、SEMA3A、FGF8和PROKR2蛋白之间具有相互作用,但与LZTR1蛋白无相互作用(图1B)。此外,Gene Ontology分析表明,FGFR1、SEMA3A、FGF8和HS6ST1影响神经元发育,FGFR1、SEMA3A和FGF8影响前脑神经元生成(图1C)。因此推测,FGFR1+HS6ST1、FGFR1+SEMA3A、FGFR1+FGF8、FGFR1+PROKR2、FGFR1+LZTR1的有害突变组合可能导致CHH发生。
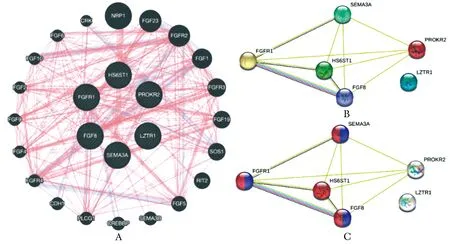
A:gene-gene interactions were predicted using genemania. Blue lines indicate co-localization. Red lines indicate physical interactions. Purple lines indicate co-expression; B:association between FGFR1 protein and other proteins. Green lines indicate text mining. Black lines indicate co-expression. The pink line indicates that the association was experimentally determined. The blue line indicates that the association was determined from a curated database; C:pathways highlighted by gene ontology analysis. Red indicates genes that affect neuron development. Purple indicates genes that affect forebrain neuron generation
在自发突变组(n=6)中均未发现其他CHH相关基因突变。
3 讨论
本研究筛查到15例FGFR1突变的CHH患者中存在15个致病性FGFR1突变。携带FGFR1突变的父/母性腺轴功能正常,但子代发生CHH。这表明来自父/母的FGFR1单基因突变不足以导致CHH,此时可能需另一个CHH相关基因突变的损伤作用积累,才会导致CHH发生。此外,Fgfr1缺失小鼠表现多样,可出现青春发育延迟,生殖能力下降,不育等。
研究提示,多个微效基因缺陷积累可能导致CHH发生[10]。不同的基因突变组合,例如FGFR1+FGFR1或GNRHR+GNRHR或PROKR2+IR17LD、FGFR1+GNRHR、FGFR1+FGF8、FGFR1+KAL1、FGFR1+NELF、PROK2+PROKR2或FGFR1+PROKR2,均可能导致疾病发生[10-11]。本研究中,遗传突变组的6例(6/9)患者存在双基因突变,为“双基因突变导致CHH发生”理论提供了更多证据。
这些基因突变会损伤GnRH神经元功能。原因如下:1)FGF8是FGFR1的主要配体。FGF8-FGFR1信号通路在调节GnRH神经元迁移和分化中发挥关键作用。因此FGFR1和FGF8突变组合会导致CHH[12]。2)Sema3a突变小鼠GnRH神经元数量减少,但单纯Sema3a杂合突变难以导致CHH。已经报道的2例同时携带FGFR1和SEMA3A突变的CHH患者[13],和本研究中病例相似。3)HS6ST1编码的蛋白,参与FGF8-FGFR1信号作用,突变影响GnRH神经元迁移。4)LZTR1突变可能导致GnRH神经元迁移障碍,引起CHH和双侧隐睾[14]。综上所述,这些基因突变引发的致病性较弱,常需要另一个CHH基因突变才会致病。
本研究存在的局限性。1)未对突变基因进行功能学验证,而只通过软件进行了预测。2)在遗传突变组中,还有3例患者未发现另一个CHH相关基因突变。这和对候选CHH致病基因认识不足有关。可能存在其他致病基因突变,协同积累损伤作用而导致疾病发生。
总之,来自生殖表型正常父/母的FGFR1突变患者,常需另一个CHH相关基因突变的协同打击才会导致CHH。研究结果拓展了对双基因突变导致CHH的机制的认识。
猜你喜欢
广西医科大学学报(2022年5期)2022-06-07
昆明医科大学学报(2022年3期)2022-04-19
中国生殖健康(2020年2期)2021-01-18
中南医学科学杂志(2019年6期)2019-12-05
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
湖南畜牧兽医(2016年3期)2016-06-05
兽医导刊(2016年12期)2016-05-17
浙江医学(2014年17期)2014-04-13
河南医学研究(2014年5期)2014-02-27
