
分光光度法测定配制酒中氰化物的不确定度评定
2023-04-02 10:05叶春常陈桂云黎颖欣胡雯艳陈韵
酿酒科技 2023年3期
叶春常,陈桂云,黎颖欣,胡雯艳,陈韵
(广州市食品检验所,广东广州 511400)
酒中氰化物主要由原料(如木薯、代用品、豆类及其他果核或混入一些野生植物)中的氰甙类配糖体在发酵过程中水解产生[1]。氰化物属于有毒物质,极少量的氰化物就会导致人或者动物中毒甚至死亡。因此,氰化物是酒类一项非常重要的安全指标[2]。
目前用于测定酒中氰化物的方法主要有分光光度法[3-4]、原子吸收法[5-6]、硝酸银滴定法[7]、气相色谱法[8-9]、离子色谱-脉冲安培检测法[10]等。国家标准方法GB 5009.36—2016《食品安全国家标准 食品中氰化物的测定》[11]中测定氰化物的方法就是分光光度法和气相色谱法。在上述的检测方法中,分光光度法是最常见的方法。
本研究参考CNAS—GL006:2019《化学分析中不确定度的评估指南》[12],通过分析分光光度法测定配制酒中氰化物的过程中影响检测结果的不确定度因素,评估该检测的不确定度,为实验室质量控制提供科学、准确的依据[13]。
1 材料与方法
1.1 材料、试剂及仪器
试验样品:市售配制酒。
试剂及耗材:酚酞、氢氧化钠、磷酸二氢钾、冰乙酸、无水乙醇(均为AR),广州化学试剂厂;磷酸氢二钠(AR),广东光华科技股份有限公司;异烟酸(AR),阿拉丁;酒石酸、氯胺T(均为AR),国药集团化学试剂有限公司;吡唑啉酮(AR),上海晶纯化学试剂厂;氰化物标准溶液(50 μg/mL),中国计量科学研究院。
仪器设备:U-3010 紫外可见分光光度计,日本日立公司;WD20A12E 数控水浴锅,美国Poly-Science 公司;C-MAG HP 10(S25)控温加热器,艾卡广州仪器设备有限公司。
1.2 试验方法
1.2.1 分析步骤
参考GB 5009.36—2016 中5.2 配制酒的方法进行处理。
1.2.2 计算公式
配制酒中氰化物(以HCN计,按100 %酒精度换算)的含量按下式计算:
式中:X为样品中氰化物含量,mg/L;X0为平均实测含量(以CN-计),μg;m 为试样体积,mL;V 为样品稀释倍数;P 为酒精度,%vol;1000 为换算系数;MHCN为氰化氢(HCN)的摩尔质量,g/mol;MCN为氰离子(CN-)的摩尔质量,g/mol。
1.2.3 测量不确定的来源分析
根据检验过程及计算公式,氰化物含量的不确定度来源及其分量如表1所示。
试样体积、实测含量和样品制备的重复性合并为整体样品测定重复性考虑。样品配制酒为液体,取样时充分摇匀,可认为试样是均匀的,具有代表性,所以样品均匀性对不确定度的影响可忽略不计。相应的不确定度来源及其分量修正如表2所示。

表2 经过修正后的不确定度来源及其分量
合成标准不确定度可用各分量的标准不确定度合成:
上式可转换为:
2 结果与分析
2.1 试样体积引入的不确定度urel(1)评定
浑浊或有色样品,取25.0 mL 试样进行蒸馏。不确定度来源为移液管的校准偏差和温度效应,校准偏差的计算可参照校准证书提供的扩展不确定度计算,温度效应根据公式(4)计算:
其中,2.1×10-4为水的膨胀系数,单位为/℃;t0为量器校准时的室温,20 ℃;t1为配制标准溶液时的室温,25 ℃。
移液管引入的相对标准不确定度按公式(5)计算:
其中,U 为移液管校准证书提供的扩展不确定度U=0.015 mL,k 为证书提供的包含因子k=2,V 为移液管的体积。
2.2 样品实测含量引入的不确定度urel(2)评定
2.2.1 标准曲线拟合引入的不确定度urel(2.1)
氰化物含量是通过最小二乘法拟合的曲线计算所得,不确定度与样品测定的次数和标准系列测定次数有关,同时和样品平均值与标准系列平均值的差值有关。
检测过程中,配制6个浓度点(0.00 μg、0.40 μg、0.80 μg、1.20 μg、1.60 μg、2.00 μg)的标准系列,每浓度点平行测定3次,制备10 个平行样,得到10 个样品测定值,用最小二乘法计算标准曲线的回归方程:y=bx+a,根据拟合的回归曲线求得被测样品的氰化物含量。测定结果见表3、表4。
根据拟合得到的曲线,按照表3、表4、公式(6)、公式(7)计算urel(2.1),相关数据填入表5中。

表3 回归曲线的吸光度计算

表5 标准曲线拟合引入的不确定度urel(2.1)计算
2.2.2 标准溶液引入的不确定度urel(2.2)评定
标准溶液引入的不确定度包括标准溶液浓度的不确定度urel(2.2.1)和标准溶液配制过程引入的不确定度urel(2.2.2),则:
2.2.2.1 标准溶液浓度的不确定度urel(2.2.1)评定
根据标准物质证书,氰化物标准溶液标准值为50.0 μg/mL,相对扩展不确定度为1 %(k=2),转换成相对标准不确定度:
2.2.2.2 标准溶液稀释过程引入的不确定度urel(2.2.2)评定
标液稀释:用单标吸管吸取2.00 mL 标准溶液至100 mL 容量瓶中,用2 g/L 氢氧化钠溶液定容,为标准使用液。2 mL 刻度吸管分别吸取0 mL、0.40 mL、0.80 mL、1.20 mL、1.60 mL、2.00 mL 标准使用液于10 mL 具塞比色管中。标准溶液稀释过程中的体积受玻璃量器的容量偏允差和实验室温度的波动影响,配制过程中使用单标吸管、容量瓶、刻度吸管、具塞比色管,将各类量器相应的数据填入表6中,参考公式(4)、(5)分别计算出各量器的相对标准不确定度。

表6 量器引入的不确定度
按B 类评定计算相对标准不确定度标准溶液稀释过程引入的不确定度urel(2.2.2):
2.2.3 仪器示值引入的不确定度urel(2.2.3)评定
检测过程使用的分光光度计的示值分辨率会对吸光度值读数产生影响,从而引入了不确定度,仪器计量证书提供的扩展不确定度U=0.3 %(k=2),由样品实测含量平均值X0代入回归方程y=bx+a 中得到平均吸光度值A=0.324,仪器示值引入的不确定度计算可按公式(12)进行。
样品实测含量引入的相对标准不确定度urel(2)为:
2.3 样品制备引入的不确定度urel(3)评定
试样进行蒸馏后定容至50 mL比色管,用2 mL单标吸管吸取2.0 mL 馏出液测定。此时不确定度来源为馏出液移取移液管urel(3.1)和定容比色管体积不确定度urel(3.2)。由公式(14)、表6得:
2.4 样品酒精度引入的不确定度评定urel(4)评定
样品氰化物含量平均值X0(以CN¯计)需经酒精度换算为试样中氰化物(以HCN计,按100%vol酒精度换算)的含量,酒精度的结果引入了不确定度。经测定,样品酒精度含量P=41.2%vol,扩展不确定度U=0.3 %vol(k=2),酒精度的相对标准不确定度为:
2.5 样品重复性测定引入的不确定度urel(5)评定
制备10 个平行样,每个样品测定2 次。测定结果如表4。
单次测量的不确定度为:
测量结果相对标准不确定度为:
2.6 氰化氢摩尔质量引入的不确定度urel(6)评定
从现行有效的IUPAC 原子量表中查得HCN 和CN-各元素的原子量和不确定度:
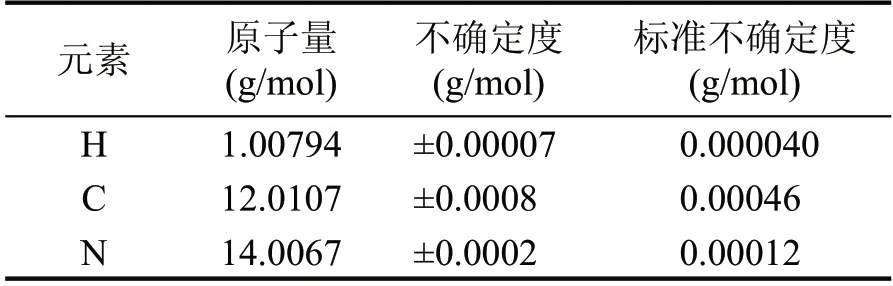
表7 HCN中各元素的原子量和不确定度
对于每一个元素来说,标准不确定度是将IUPAC 所列不确定度做为矩形分布的边界计算得到的。因此相应的标准不确定度等于查得数值除以。
HCN的摩尔质量为:
相对标准不确定度为:
2.7 氰离子摩尔质量引入的不确定度urel(7)评定

表8 各分量的不确定度汇总
CN-的摩尔质量为:
相对标准不确定:
2.8 合成标准不确定度及扩展不确定度计算
2.8.1 合成标准不确定度
将上述数据urel(1)—urel(7)代入公式(2)得到被测样品含量的合成相对标准不确定度uc,rel(X),再按公式(22)转换成合成标准不确定度uc(X):
2.8.2 扩展不确定度
测量结果符合正态分布,取置信概率p=95%,包含因子k=2,则扩展不确定度U为:
2.9 结果报告
样品中氰化物的含量为(2.5±0.1)mg/L,k=2。
3 结论
本研究采用分光光度法对配制酒中的氰化物含量进行检测,分析评估检测过程的各个不确定度来源及其大小。结果表明,实测含量、测定重复性引入的不确定度最大,样品制备和酒精度引入的不确定度次之,试样体积、氰化物和氰离子的分子量引入的不确定度最小。因此,在检测过程中,标准溶液的配制要准确,仪器应该严格按要求及时校准并保持良好的状态,规范检测过程中的操作,才能更好提高检测的准确度。
猜你喜欢
山东冶金(2022年3期)2022-07-19
供水技术(2021年3期)2021-08-13
广西蚕业(2021年2期)2021-07-15
天津化工(2019年6期)2019-12-10
食品与发酵工业(2019年1期)2019-01-29
现代园艺(2017年21期)2018-01-03
电镀与环保(2017年3期)2017-06-23
西藏科技(2016年9期)2016-09-26
食品工程(2015年3期)2015-12-07
表面工程与再制造(2014年2期)2014-02-27
