
CRISPR/Cas系统的核酸检测及其在食品安全快速检测中的应用
2023-03-23 06:22:46翟百强王洪涛徐瑗聪
中国食品学报 2023年2期
翟百强,程 楠,王洪涛,徐瑗聪
(1 北京工业大学环境与生命学部 北京 100024 2 河南省铁路食品安全管理工程技术研究中心 郑州 450052 3 中国农业大学食品科学与营养工程学院 北京 100083)
食品安全关乎民生,快速、精准的检测能够为食品安全事件提供有效的科学预警。生物传感器检测技术能够将未知靶标有效转化成可测量的信号,成为食品安全快速检测领域的佼佼者。核酸传感器依托核酸中间体承载信号的识别、放大和转导的作用,在食源性致病菌、农药残留、重金属检测、生物掺伪等[1-3]检测中表现优异,有效缩短了检测时间,提高了检测特异性及灵敏度。2020年诺贝尔化学奖揭晓后,CRISPR/Cas(Clustered regularly interspaced short palindromic repeats,CRISPR;CRISPR-associated protein,Cas)系统因优异的基因编辑能力、独特的靶点识别方式、极高的酶活力等特性广为人知,在核酸检测领域表现出巨大的应用价值[4-6]。由其构建的核酸传感器也成为食品安全快速检测平台的研究热点。本文就基于CRISPR/Cas系统的研究进行综述,分析不同CRISPR/Cas 系统结合核酸技术在不同靶标检测中的应用情况,展望CRISPR/Cas 核酸传感器在食品安全快速检测领域的未来研究方向,以促进其在食品安全检测中的进一步应用。
1 CRISPR/Cas 系统概述
CRISPR-Cas 系统即成簇规律间隔的短回文重复序列以及CRISPR 相关蛋白系统,是一种获得性免疫系统,存在于众多古细菌和细菌中[7]。根据系统依赖的Cas 蛋白的不同可以分成2 大类[8]:第1 类Cas 蛋白是多亚基蛋白复合物,发挥活性需要依靠多个效应蛋白协同作用,第2 类Cas 蛋白是独立蛋白,发挥活性仅依靠单一效应蛋白。根据Cas 蛋白的基因结构及重复间隔序列结构不同可以分为6 类(Ⅰ~Ⅵ型),其中Ⅰ型、Ⅲ型和Ⅳ型属于第1 类系统,Ⅱ型、Ⅴ型和Ⅵ型属于第2 类系统[8-9]。CRISPR/Cas系统的作用机制包括3 个阶段:获取、表达和干扰。首先将外源基因整合到CRISPR 基因座2 个重复序列中间,获得间隔序列;其次基因座转录成前体crRNA,随后加工为成熟crRNA(CRISPR RNA);最后crRNA 与Cas 蛋白复合,发挥核酸酶活性对外源基因进行切割[9]。基于CRISPR/Cas系统的检测技术则是依据作用机制,根据检测靶标序列设计向导RNA(Singleguide RNA,sgRNA),当其与特异性靶标识别配对后,激活Cas 蛋白的切割活性,通过切割产物的分析实现靶标的检测。目前的研究主要集中在Cas9、Cas12、Cas13 以及Cas14 这4 种蛋白(图1),其中基于Cas9 的检测侧重于sgRNA 的设计,通过特异性识别靶序列实现多重检测;基于Cas12、Cas13 及Cas14 的检测则重点依赖Cas 蛋白激活后的切割活性。
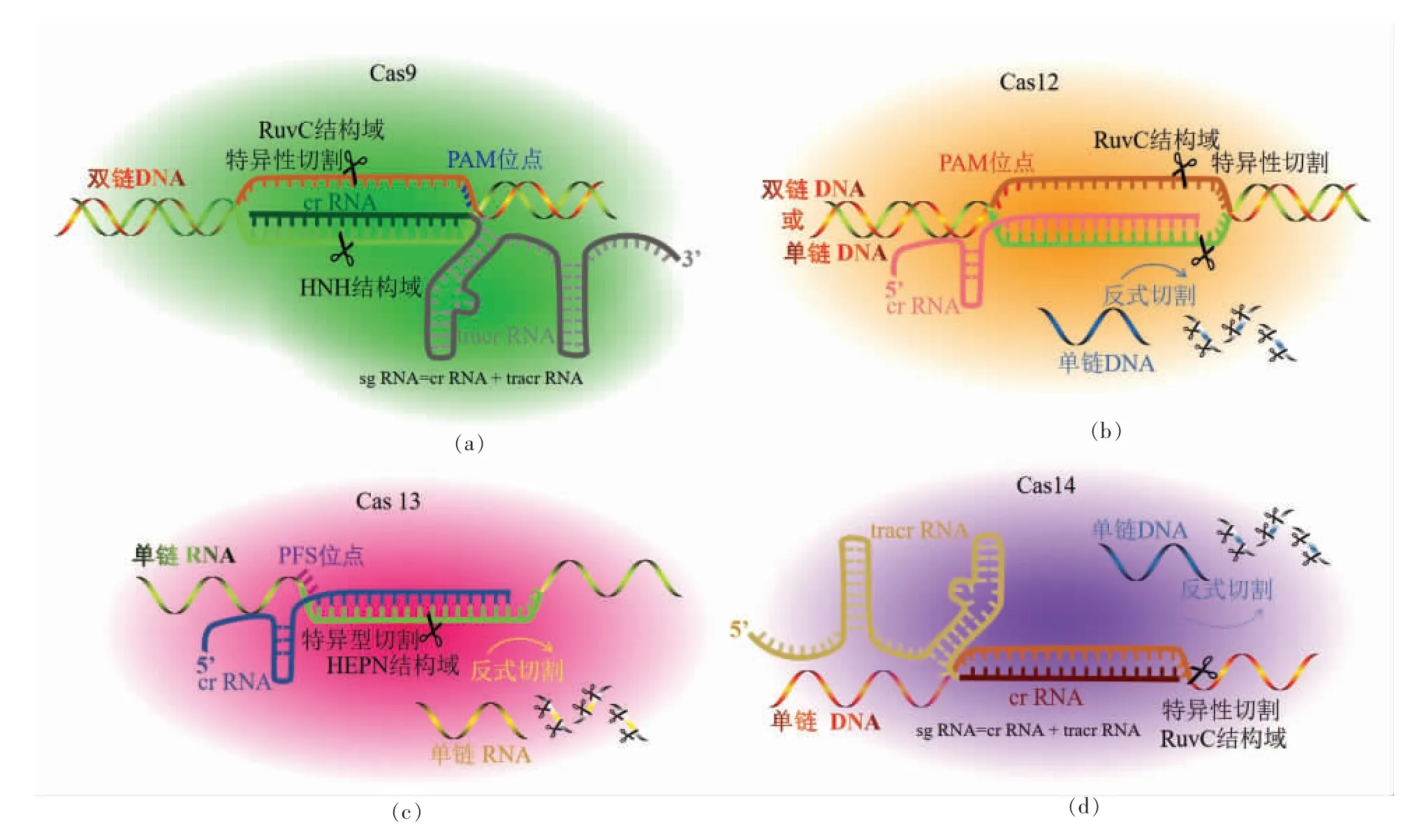
图1 CRISPR/Cas9(a)、Cas12(b)、Cas13(c)及Cas14(d)基本组成及作用原理图Fig.1 Fundamental components and mechanism of CRISPR/Cas99(a),Cas12(b),Cas13(c),and Cas14(d)systems
1.1 CRISPR-Cas9
Cas9 蛋白是最为典型的2 类Ⅱ型CRISPR 系统效应蛋白,是RNA 引导和dsDNA(Doublestrand DNA)靶向的核酸内切酶,含有HNH 和RuvC 2 个核酸酶结构域,其中HNH 只有1 个核酸酶区域,RuvC 含有3 段核酸酶区域[10]。CRISPRCas9 系统需要Cas9、crRNA 和tracrRNA(Transactivating RNA)协同发挥作用,其中crRNA 与tracrRNA 合并设计形成一条sgRNA,当Cas9 与crRNA 部分结合后,能够识别含有PAM(Protospacer adjacent motif)位点(5'-NGG)的靶标dsDNA,crRNA 与靶标dsDNA 中的互补链形成RNADNA 异源双链结构,随后Cas9 的两个核酸酶结构域活性被激活,进而切断靶标dsDNA[11]。后有研究发现单独添加小段PAMmer 序列,与ssDNA 靶标形成含有PAM 序列的部分dsDNA 后,Cas9 依然能够发挥切割活性对ssDNA 靶标进行切割[12]。此外研究通过对Cas9 的HNH 和RuvC 结构域进行突变,得到没有活性的dCas9(Nuclease-deactivated Cas9)蛋白,虽能够识别靶标dsDNA,但是没有核酸酶切割活性[13]。
1.2 CRISPR-Cas12
Cas12 蛋白属于2 类Ⅴ型CRISPR 系统效应蛋白,也是RNA 引导和DNA 靶向的核酸内切酶,只含有RuvC 核酸酶结构域[8]。CRISPR-Cas12 系统中,Cas12 蛋白能够在crRNA 的引导下识别含有PAM 位点(5'-TTTN)的靶标dsDNA,或者识别不需要PAM 位点的ssDNA(Single-strand DNA)靶标序列,形成三链或者双链复合体,随后激活RuvC 的核酸内切酶活性,切割特异性靶标dsDNA或者靶标ssDNA(也称作cis 切割),同时还激活了反式切割活性,能够切割其它非特异性ssDNA(也称作trans 切割)。其中反式切割活性的激活需要一个必备条件,就是要求靶标模板序列中至少有15 个碱基与crRNA 互补。由于dsDNA 靶标中非互补链有助于提升“互补链/crRNA/Cas12 蛋白”的稳定性,因此dsDNA 靶标激活的反式切割能力要远远高于ssDNA 靶标。Cas12 蛋白的家族成员包括Cas12a~Cas12f,但研究多集中在Cas12a 和Cas12b。Cas12a 又称为Cpf1 蛋白,Cas12b 又称为C2c1 蛋白,两者的区别在于前者的crRNA 成熟不需要tracrRNA 参与,而后者需要,但是两者都同时具备核酸内切酶活性和反式切割活性[14-16]。
1.3 CRISPR-Cas13
Cas13 蛋白属于2 类Ⅵ型CRISPR 系统效应蛋白,是RNA 引导和RNA 靶向的核酸内切酶,含有两个HEPN 核酸酶结构域[8]。CRISPR-Cas13 系统中,Cas13 蛋白本身能够对crRNA 的成熟进行加工处理,不需要tracrRNA 参与,随后在crRNA的引导下识别含有PFS(protospacer-flanking sequence)位点的(3'-A 或U 或C)的RNA,形成双链RNA 复合物促使HEPN1 和HEPN2 两个结构域相互靠近,从而激活HEPN 核酸酶的核酸内切酶活性,切割特异性靶标RNA,同时还激活了反式切割活性,能够非特异性切割其它非靶标RNA[17-18]。Cas13 蛋白的家族成员包括Cas13a~Cas13d,其中关于Cas13a 的研究较多,不同的Cas13 对切割探针具有不同的偏好性。Cas13a 又称为C2c2 蛋白,通过对核酸酶区域的残基突变可得到没有核酸酶活性dCas13a,能够结合靶标RNA 却没有切割性能[19]。
1.4 CRISPR-Cas14
Cas14 蛋白属于2 类Ⅴ型CRISPR 系统效应蛋白,是RNA 引导和ssDNA 靶向的核酸内切酶,含有RuvC 核酸酶结构域。CRISPR-Cas14 系统与Cas9 系统相似,需要Cas 蛋白、crRNA 和tracr-RNA 协同发挥作用,不同的是Cas14 的识别不需要PAM 位点。其切割特性与Cas12 相似,与Cas13的区别在于识别的靶标不同,Cas14 专一识别靶标ssDNA 后激活RuvC 核酸酶的核酸内切酶活性,切割特异性靶标ssDNA,同时还激活了反式切割活性,能够非特异性切割其它非靶标ssDNA[20]。由于CRISPR/Cas14 系统对crRNA 和ssDNA 靶标的错配容忍度很低,成功突破了ssDNA 靶标精准识别的短板,成为了CRISPR 系统巨大的进步。其家族成员包括Cas14a~Cas14c,是目前最小的第2 类CRISPR 效应蛋白[21]。后有研究报道,Cas14 是属于Cas12f 的亚类,其中Cas14a 属于Cas12f1,Cas14b属于Cas12f2,而Cas14c 属于Cas12f3[22]。
2 CRISPR/Cas 系统与核酸扩增技术的结合应用
不同的CRISPR/Cas 系统识别不同类型的靶标,在检测过程中直接识别则存在靶标浓度低,基质干扰等问题,一般需要通过核酸扩增技术对靶标进行放大,再利用产物作为新的靶标与CRISPR/Cas 系统结合建立相应的核酸传感器检测平台。不同的核酸扩增技术能够生产不同类型的靶标产物,两者之间的组合能够充分发挥CRISPR/Cas系统的检测功能,目前已组装成多种类型的CRISPR/Cas 核酸检测平台并广泛应用于各大检测领域。各平台之间的差异和改进多集中在5 个方面:操作步骤(扩增和检测两步反应的合并)、特异性(碱基突变的识别)、过程污染(闭管反应避免交叉污染)、检测通量(单重升级到多重检测)以及结果输出(实现裸眼可视化)。核酸扩增技术根据温度依赖程度可以分为恒温扩增和变温扩增,根据产物类型可以分为双链产物扩增和单链产物扩增,其中能够生成双链产物的恒温扩增技术应用较多,典型代表是重组聚合酶扩增技术(Recombinase polymerase amplification,RPA),有经典的SHERLOCK、DETECTR 等检测平台。
2.1 CRISPR/Cas 系统结合NASBA
NASBACC(NASBA/CRISPR cleavage)检测平台是CRISPR/Cas 系统与核酸扩增技术的首次结合应用。核酸序列依赖扩增(Nucleic acid sequence-based amplification,NASBA)是以RNA为模板,依赖AMV 反转录酶、Rnase H 酶和T7RNA 聚合酶的等温扩增技术,其中间产物为DNA 双链。NASBACC 方法中,以寨卡病毒的RNA为靶标进行NASBA 扩增,中间产物DNA 转录得到RNA,然后通过小立足点置换激活传感器上的颜色反应,通过黄色和紫色的区分实现基因分型。方法的巧妙之处在于,CRISPR/Cas9 系统中cr-RNA 选取的PAM 序列恰好是两种寨卡病毒的基因差异片段。美国寨卡病毒通过NASBA 扩增后能够得到含有“CGG”的dsDNA 产物,与crRNA 进行和互补配对,并激活Cas9 蛋白的切割活性,将ds-DNA 切断,随后转录只能得到小段RNA,无法激发后续反应而呈现出黄色结果。非洲寨卡病毒存在碱基突变,扩增后得到的是含有“CAG”的dsDNA 产物,由于PAM 序列的突变,无法激活Cas9的切割活性,此时dsDNA 能够转录得到完整的RNA,通过小立足点置换反应激活后续的生化反应呈现紫色结果,从而实现对寨卡病毒的精准分型鉴定[23]。
2.2 CRISPR/Cas 系统结合RPA
SHERLOCK(Specific high-sensitivity enzymatic reporter unlocking)[24]和DETECTR(DNA endonuclease targeted CRISPR trans reporter)[14]是CRISPR/Cas 系统结合RPA 扩增技术的2 个经典平台。RPA 是以DNA 为模板,依赖重组聚合酶、单链结合蛋白和链置换DNA 聚合酶的等温扩增技术,其扩增产物是DNA 双链。
张锋团队建立的SHERLOCK 平台,是基于CRISPR/Cas13a 与RPA 进行结合,整体包括2 个步骤。先由RPA 扩增生成dsDNA 产物,由于Cas13a 识别的是RNA 靶标,因此需要对dsDNA产物进行转录得到RNA 产物,随后与crRNA 结合并激活Cas13a 的反式切割活性,切割荧光淬灭的RNA 探针,通过荧光的变化实现了寨卡病毒和登革病毒的区别检测、致病菌的检测、人类基因组分型以及单碱基突变检测等。如果靶标是RNA,则需要先进行反转录再完成RPA 扩增[24]。不同的Cas13 蛋白对切割探针具有不同的偏好性,该团队基于该特性进一步建立了SHERLOCKv2 平台,通过 PsmCas13b、LwaCas13a、CcaCas13b 和 Cas12a对不同荧光探针的切割,实现了四重检测,后续通过结合CRISPR 相关酶Csm6 进一步提升灵敏度,并应用于侧流层析试纸条实现可视化检测[25]。SHERLOCK 中RPA 扩增需要对靶标进行基因组提取,为了简化核酸的提取步骤,该团队再次建立HUDSON(Heating unextracted diagnostic samples to obliterate nucleases)平台。通过加热和化学处理释放核酸,并使核糖核酸酶失活,从而实现寨卡病毒和革登热病毒的SHERLOCK 快速检测[26]。Arizti-Sanz 等[27]基于上述研究,组合SHERLOCK和HUDSON 建立了SHINE(SHERLOCK and HUDSON integration to navigate epidemics)平台,通过体系优化实现了两步法检测的一体化,成功用于SARS-CoV-2 的免提取快速精准检测。这一系列检测平台虽然具有良好的特异性和灵敏度,但是由于依赖的是Cas13 蛋白,因此需要增加反转录过程,一定程度上增加了操作的繁琐性。
DETECTR 平台[14]是基于CRISPR/Cas12a 与RPA 结合的原理建立,含有PAM 序列的dsDNA产物与crRNA 结合并激活Cas12a 的反式切割活性,切割荧光淬灭的ssDNA 探针,通过荧光的变化进行结果判定。利用“TTTC”PAM 序列后的6 个碱基差异,成功实现人乳头瘤病毒HPV16 和HPV18 的精准快速检测。随后又基于Cas14a 建立了Cas14-DETECTR 平台,用于人类眼球颜色基因HERC2 中单核苷酸多态性(Single nucleotide Polymorphism,SNP)的分型鉴定。由于Cas14a 识别的是ssDNA 靶标,反应中先由硫代磷酸化引物(只有一条引物进行标记)进行RPA 扩增生成单标记dsDNA 产物,随后利用T7 核酸外切酶对没有标记的链进行水解得到ssDNA 产物,随后与crRNA 结合并激活Cas14a 的反式切割活性,对荧光淬灭的ssDNA 探针进行切割。鉴于CRISPR/Cas14 系统对crRNA 和ssDNA 靶标的错配容忍度很低,能够在没有PAM 序列的条件下有效区分单碱基突变,该方法成功实现蓝眼球和棕眼球的SNP 检测,相比之下Cas12a 无法区分[20]。为了简化反应流程,实现扩增和检测一体化,Wang 等[28]合并DETECTR 建立了Cas12aVDet(Cas12a-based visual detection)平台,将所有试剂整合在一个管中,Cas12a 附着在管壁上,RPA 扩增后通过离心将Cas12a 与之混合进而完成切割,最后通过蓝光激发实现裸眼判定。该方法能够在与DETECTR保持相同灵敏度的条件下,成功将检测时间从2 h 缩短至0.5 h,并且全程闭管反应有效避免了交叉污染。
除了以上经典检测平台之外,还有许多关于CRISPR/Cas 系统和RPA 的联合应用,大部分致力于特异性、灵敏度及便携性上的升级。例如基于Cas12b 的CDetection(Cas12b-based DNA detection)用于HPV 检测,灵敏度优于DETECTR。同时利用单碱基错配的tgRNA(Tuned guide RNA)提高检测特异性,实现血型SNP 分型检测[29]。例如基于Cas12a 的AIOD-CRISPR(All-In-One Dual CRISPR-Cas12a)用于SARS-CoV-2 检测,利用产物在上下游设计两条crRNA,通过提升Cas12a 的利用率来提升检测灵敏度[30]。例如基于Cas13a 的DESCS(Dual methylation sensitive restriction endonucleases coupling with an RPA -assisted CRISPR/Cas13a System)用于SEPT9 基因的甲基化检测,利用限制性内切酶对甲基化位点的敏感性来调控后续RPA 反应,存在甲基化位点则限制性内切酶不产生切割,此时靶基因连续就能继续发生RPA 扩增,进而完成Cas13a 的有效切割,结合侧流层析试纸条实现可视化检测[31]。
2.3 CRISPR/Cas 系统结合PCR
聚合酶链式反应(Polymerase chain reaction,PCR)是最为传统的核酸扩增技术,属于变温扩增,产物为dsDNA。HOLMES(one-hour low-cost multipurpose highly efficient system)是基于CRISPR/Cas12a 与PCR 结合的经典平台,较SHERLOCK 而言更适合DNA 靶标检测。通过含有PAM 序列的dsDNA 产物与crRNA 结合并激活Cas12a 的反式切割活性,通过探针切割反应中荧光的变化完成。对于24nt crRNA 而言,HOLMES能够识别PAM 序列以及PAM 后1~7 位碱基中单碱基突变,但是无法区分8~18 位碱基中的突变;对于16nt 和17nt crRNA 而言,HOLMES 能够识别PAM 后1~16 位碱基中的突变。根据这一特性,在SNP 位点附近设计插入PAM 序列的PCR 引物,提升对SNP 检测的特异性和灵敏度。HOLMES检测平台中,核酸扩增技术并不局限于PCR,还可以采用其它等温扩增技术[32]。多重PCR 实现了单一检测向高通量检测的进阶,Wang 等[33]将多重PCR 与CRISPR/Cas12a 结合建立了多重检测平台,针对不同的靶标设计不同的crRNA 序列,并将crRNA 分别装载入不同的反应孔中,最后通过荧光的变化实现五重检测。普通PCR 扩增耗时长达2 h 左右,快速PCR 能够显著缩短反应时间至5 min 内,Wang 等[34]将快速PCR 与CRISPR/Cas12a 结合建立了CRISPCR(CRISPR & rapid PCR in single capillary),将快速PCR 反应试剂与Cas12a 切割反应试剂分别置于反应管的上下两部分,扩增后再混匀试剂进行切割,紫外照射后实现裸眼判定。该方法与Cas12aVDet 有异曲同工之妙,但能够在10 min 内完成整体检测,效率上更胜一筹。
2.4 CRISPR/Cas 系统结合LAMP
环介导等温扩增技术(Loop mediated isothermal amplification,LAMP)是经典的恒温扩增技术,能在短时间内生成大量的dsDNA 产物,并且不依赖于热循环仪器。为了简化操作和避免反应交叉污染,同一团队在HOLMES 的基础上,将 CRISPR/Cas12b 和 LAMP 结合 建 立 了HOLMESv2 平台,用于DNA 和RNA 靶标的检测,并有效区分SNP。在LAMP 产物中选取含有PAM位点的片段作为检测靶标,与crRNA 互补配对后激活Cas12a 的反式切割活性,根据荧光的变化进行结果分析。该检测平台中的核酸扩增技术并不局限于LAMP,也可以是PCR,或者是能够生成单链产物的非对称PCR。该方法还可以用于检测基因甲基化,利用酸性亚硫酸盐处理靶基因,甲基化的胞嘧啶保持不变,未甲基化的胞嘧啶变成尿嘧啶,经过PCR 扩增后,只有甲基化的产物与cr-RNA 配对结合,产生荧光的变化[35]。结合LAMP 的其它应用也比较多,例如TB-QUICK 技术中,利用LAMP 进行核酸扩增,基于CRISPR/Cas12b 的cis切割活性实现结核杆菌的检测[36];CAL-LAMP(CRISPR/Cas12a assisted ligation-initiated LAMP)技术中,利用靶标连接发夹引物进而触发LAMP反应,基于CRISPR/Cas12a 的trans 切割活性实现microRNA let-7a 的检测[37]。虽然LAMP 应用十分广泛,但是污染问题一直是限制该技术的最大弊端,为了避免交叉污染引起的假阳性,Qian 等[38]建立了尿嘧啶-LAMP,采用dUTP 代替dTTP,因此得到的是带有dUTP 的扩增产物。同时发现Cas12bde PAM 序列可以由“TTTN”更改为“UUUN”,且不影响各种切割活性。因此结合尿嘧啶糖基化酶(UDG)、尿嘧啶-LAMP 和CRISPR/Cas12a建立了ULC(UDG-LAMP-CRISPR)检测平台。先用UDG 进行酶解消化,去除体系中残存的dUTP扩增产物,再进行扩增和后续的切割反应,有效避免了反应污染问题。ULC 方法虽然有效,但是一定程度上增加了操作的复杂性,同样是为了解决污染问题,Bao 等[39]基于CRISPR/Cas9 建立了CUTLAMP(Contamination-free loop-mediated isothermal amplification),通过在LAMP 内引物中添加PAM 序列,使得扩增产物中含有重复的PAM 序列,在Cas9/crRNA 的识别下能够将产物进行切割降解,从而避免污染问题,有效杜绝假阳性反应。LAMP 技术是基于DNA 扩增的检测方法,针对RNA 靶标,则需要通过反转录建立RT-LAMP,随后将其与CRISPR/Cas12a 系统进行结合,建立了iSCAN(in vitrospecific CRISPR-based assay for nucleic acids detection)检测平台,搭载侧流层析试纸条实现SARS-CoV-2 的可视化检测[40];建立了opvCRISPR(a one-pot visual reverse transcription(RT)-LAMP-CRISPR)检测平台,通过蓝光激发实现SARS-CoV-2 的目视分析,具有良好的特异性灵敏度以及准确性[41]。
2.5 CRISPR/Cas 系统结合单链扩增技术
对于原始靶标信号来说,核酸扩增能够有效放大靶标浓度,进而激活Cas 蛋白的活性。像RPA、LAMP 等这些扩增方法产物都是dsDNA,能够直接与CRISPR 系统进行结合,但是还有许多核酸扩增技术的产物是ssDNA,与CRISPR 系统结合时具有一定的选择性,例如CRISPR/Cas12 和CRISPR/Cas14 能够识别单链靶标,则能直接结合应用,而CRISPR/Cas9 与之结合则需要添加互补链组装成双链靶标。
滚环扩增技术(Rolling circle amplification,RCA)是单链扩增中研究较多的一种方法,能够在短时间内生产大量连续重复的ssDNA 产物。RACE(RCA-assisted CRISPR/Cas9 cleavage)检测平台是基于RCA 扩增和CRISPR/Cas9 系统联合建立的,整体包括扩增和检测两个步骤。以靶标为引物,对含有PAM 序列的环模板进行RCA 扩增,产生含有大量重复靶标序列的单链产物;添加荧光淬灭的互补探针组装成双链产物,与crRNA 识别配对后激活Cas9 蛋白的切割活性,对双链产物进行切割,导致探针断裂而释放荧光,通过荧光的变化实现对miR-21 的检测[42]。Xu 等[43]利用适配体进行靶标转导,基于RCA 和CRISPR/Cas12a 建立了针对耐甲氧西林金黄色葡萄球菌的快速检测方法。选择菌体表面的A 蛋白和PBPa2 蛋白的适配体A 和B,其中适配体A 与磁珠偶连,适配体B用互补链进行封闭,当靶标菌体与两条适配体混合时,通过磁分离能够将“适配体A-菌体-适配体B”进行分离,剩下封闭链用于RCA 扩增,随后与crRNA 互补后激活Cas12a 蛋白的反式切割活性,利用荧光变化完成检测。Qing 等[44]利用RCA 作为一种转导扩增,结合CRISPR/Cas12a 实现不同类型靶标的通用检测。扩增部分,在环模板中设计特定的一段通用序列,根据靶标改变引物结合区序列,RCA 扩增后都能够得到含有重复通用互补序列的产物,而通用互补序列能够与crRNA 互补后激活Cas12a 蛋白的反式切割活性。检测部分,连接氧化石墨烯电极,切割探针断裂后,无法与亚甲基蓝标记的报告探针互补配对,此时的报告探针被氧化石墨烯吸附,引起电信号升高,反之切割探针与报告探针形成双链,无法被氧化石墨烯吸附,则电信号降低。通过RCA 的扩增转导、Cas12a 的反式切割以及电化学传感器的分析,实现了疾病相关核酸以及小分子的通用检测。
除RCA 应用较多之外,还有一类依赖酶切反应的单链扩增技术,其中链置换扩增技术(Strand displacement amplification,SDA)是典型代表。Wang 等[45]利用SDA 结合CRISPR/Cas12a 实现了PNKP 的超灵敏检测。发夹底物末端的磷酸基团在T4 PNKP 的作用下转化成羟基,随后发生SDA 反应,在链置换酶的作用下生成dsDNA,进而在切克内切酶的作用下生成ssDNA,两者都是Cas12a/crRNA 的靶标,进而实现对荧光淬灭探针的切割。TITAC-Cas(Target-induced transcription amplification to trigger the trans-cleavage activity of Cas13a)检测平台是基于RNA 聚合酶介导的SDA和CRISPR/Cas13a 的联合,用于碱性磷酸酶ALP的检测。磷酸基团修饰的T7 启动子序列与转录模板形成双链底物,此时T7 启动子序列能够被外切酶水解。当存在ALP 时,磷酸基团被转化成羟基从而使得T7 启动子序列得到保护,进而在T7 RAN 聚合酶的作用下生成RNA,与crRNA 结合后激活Cas13a 的反式切割活性,通过切割探针荧光的变化完成检测[46]。指数扩增技术(Exponential amplification reaction,EXPAR)是以单链靶标为引物进行扩增的SDA 反应。CAS-EXPAR(CRISPR/Cas9 triggered exponential amplification method)检测平台是基于EXPAR 和CRISPR/Cas9的联合,与以往的不同是,该平台是由CRISPR 激发EXPAR 扩增。ssDNA 靶标与PAMmer 小片段以及Cas9/sgRNA 混合后,形成含有PAM 序列的ss-DNA-dsDNA-Cas9/sgRNA 复合物,激活Cas9 蛋白活性对ssDNA 靶标进行切割,形成的产物能够作为EXPAR 的引物触发后续的扩增反应,对单核增生李斯特菌检测具有良好的特异性和灵敏度[47]。Cas-G4EX(a CRISPR/Cas9 system mediated G4-EXPAR)检测平台是基于EXPAR、G4 核酸酶和CRISPR/Cas9 的联合,用于ssRNA 和ssDNA 的方法学检测。与CAS-EXPAR 前半部分原理相似,单链靶标与PAMmer 小片段和Cas9/sgRNA 形成复合物,切断单链靶标为EXPAR 提供引物。EXPAR扩增模板中设计有两个切割位点,因此一级产物能够再次触发二次扩增,终产物是富含鸟嘌呤的单链核酸,能够在氯高铁血红素的作用下折叠成G 四面体,并呈现出类过氧化物酶活性,催化底物ABTS 发生颜色变化[48]。
2.6 CRISPR/Cas 系统结合核酸免扩增技术
对于低浓度靶标而言,核酸扩增虽能够显著放大原始信号,促进Cas 蛋白活性最大化,但也存在一些弊端,例如需要引物设计、需要扩增仪器、可能造成污染等等,因此许多免核酸扩增的技术与CRISPR/Cas 系统也开始进行结合应用。Choi等[49]利用双纳米金传感器,结合CRISPR/Cas12a实现了癌症核酸标志物的免扩增直接检测。20 nm 金颗粒上连接有7 nm 长的ssDNA 荧光探针,60 nm 金颗粒上连接有9 nm 长的ssDNA 探针,两条探针通过碱基配对组装后,探针的荧光被60 nm 金颗粒淬灭。根据核酸靶标设计crRNA,不存在靶标则无法激活Cas12a 的活性。当单链核酸靶标存在时,能够与crRNA/Cas12a 结合并激活切割活性,将探针的单链部分切断,致使荧光恢复,20 nm 金颗粒则能进一步增强荧光,通过荧光的变化能实现对癌症核酸标志物靶标的有效灵敏检测。Li 等[50]利用金属核酸酶的切割特性,结合CRISPR/Cas12a 实现重金属的超灵敏检测。根据铅离子的金属核酸酶的底物链设计crRNA,由于序列封闭使其无法与crRNA 结合,当铅离子存在时,能够切割底物链,释放游离的ssDNA,进而与Cas12a/crRNA 结合并激活反式切割活性,通过荧光的变化实现铅离子的直接检测。Niu 等[51]利用适配体的生物功能,结合CRISPR/Cas12a 建立Molecular Radar(Random molecular aptamer-dependent CRISPR-assist reporter)检测平台,实现小分子物质的直接检测检测。根据小分子物质的适配体设计crRNA,适配体与Cas 12a/crRNA 结合并激活反式切割活性,荧光强度发生变化。当靶物质存在时,能够与适配体结合引起适配体空间构象的变化,导致其无法与crRNA 结合,也就不存在反式切割。体系中靶物质浓度与荧光强度成反比,通过的荧光强度能够实现小分子靶标的灵敏检测。Sha 等[52]将CRISPR/Cas13a 和CRISPR/Cas14a 进行串联,实现了microRNA 的超灵敏直接检测。利用microRNA 作为靶标,与Cas13a/cr-RNA 结合并激活反式切割活性,将发夹底物的环单链进行切割,随后在小立足点的作用下与Cas14a/crRNA 互补结合并激发反式切割活性,切割体系中荧光淬灭的探针,通过荧光的变化实现microRNA 的免扩增检测。
3 CRISPR/Cas 核酸传感器在食品快速检测中的应用
核酸传感器是利用核酸贯穿整体检测,实现“靶标信号-核酸信号-化学信号” 的层层转化,CRISPR/Cas 核酸传感器则是在核酸传感器的基础上,将CRISPR/Cas 系统引入其中,完成整体检测信号的识别、放大与转导,显著提高检测灵敏度和特异性。目前,CRISPR/Cas 核酸传感器已逐步应用于食品安全的快速检测,例如食源性致病微生物、生物毒素、金属离子、生物技术食品、肉类掺假以及非法添加等方面。
3.1 食源性致病菌的检测
食源性致病菌引发的食品安全事件长居榜首。致病菌的CRISPR/Cas 核酸传感器检测可分为两大类(图2):第一,通过增菌后提取基因组,结合核酸扩增技术和Cas 蛋白的切割能力建立核酸传感器检测;第二,结合适配体实现致病菌向核酸的转化,随后直接或者间接与CRISPR/Cas 系统结合,建立相应的核酸传感器,实现免培养免提取检测。Peng 等[53]基于核酸扩增和CRISPR/Cas12a 建立了CRISPR/Cas-荧光传感器,用于金黄色葡萄球菌(Staphylococcus aureus)的检测。通过基因组提取进行PCR 扩增,随后将扩增产物与Cas12a/crRNA 结合,利用反式切割活性切割荧光淬灭探针,通过荧光强度的变化完成金黄色葡萄球菌的检测,最低检测限为103CFU/mL。Mukama 等[5]基于核酸扩增、CRISPR/Cas12a 和试纸条技术建立了CRISPR/Cas-侧流层析传感器,用于铜绿假单胞菌(Pseudomonas aeruginosa)的检测。通过基因组提取进行LAMP 扩增,随后将扩增产物与Cas12a/crRNA 结合,荧光淬灭探针作为连接体能够将纳米金固定在试纸条的检测线上。当探针在反式切割活性作用下被切断后,纳米金则无法被固定在检测线上,通过检测线的有无实现了铜绿假单胞菌的定性检测。Li 等[54]基于核酸扩增、CRISPR/Cas12a 和电化学技术建立了CRISPR/Cas-电化学传感器,用于单核细胞增生李斯特菌(Listeria monocytogenes)的检测。通过基因组提取进行RAA 扩增(原理与RPA 相同),随后将扩增产物与Cas12a/crRNA 结合,利用反式切割活性将电极表面ssDNA 探针切断引发电信号变化,实现了单核增生李斯特菌的检测,最低检测限为26 CFU/mL。Gootenberg 等[25]基于核酸扩增和CRISPR/Cas13 建立了CRISPR/Cas-双重荧光传感器,用于金黄色葡萄球菌和铜绿假单胞菌的检测。通过基因组提取进行多重RPA 扩增,随后将扩增产物反转录后与Cas13a/crRNA 和Cas13b/crRNA 结合,根据Cas13 蛋白对切割探针的偏好性,采取Cas13a 切割poly U 探针,Cas13b 切割poly A 探针,通过两种荧光信号强度的变化,实现了金黄色葡萄球菌和铜绿假单胞菌的同时检测。Shen 等[55]基于适配体、核酸扩增和CRISPR/Cas13a 建立了CRISPR/Cas-荧光传感器,用于肠炎沙门氏菌(Salmonella Enteritidis)的免培养检测。利用“适配体-T7 启动区-引物结合区”设计发夹探针,探针中的适配体能够有效识别靶标并与之结合,通过空间构象的变化释放出引物区,结合引物后在DNA 聚合酶的作用下形成dsDNA,进而在T7 RNA 聚合酶的作用下生成ssRNA,与Cas13a/cr-RNA 结合后激活反式切割活性,通过荧光强度的变化实现肠炎沙门氏菌的检测,最低检测限为1 CFU/mL。Li 等[50]基于适配体和CRISPR/Cas12a 建立了CRISPR/Cas-荧光传感器,用于鲍曼不动杆菌(Acinetobacter baumannii)的免培养免扩增检测。利用磁珠探针进行菌体捕获,探针中的适配体与靶标结合后释放互补链,而互补链能够结合Cas12a/crRNA 激活反式切割活性,通过荧光强度的变化实现鲍曼不动杆菌的检测,最低检测限为3 CFU/mL。

图2 食源性致病菌的CRISPR/Cas 核酸传感器检测流程示意图Fig.2 Schematic diagram of CRISPR/Cas nucleic acid biosensors detection process for foodborne pathogens
3.2 生物毒素的检测
生物毒素是由动物、植物、微生物产生的有毒有害物质,引发食品安全事件较多的是微生物分泌的毒素。由于毒素属于非核酸物质,所以在利用CRISPR/Cas 核酸传感器检测时大多需要采用适配体进行核酸转化(图3)。Abnous 等[56]基于适配体、CRISPR/Cas12a、和纳米金建立了CRISPR/Cas-比色传感器,用于黄曲霉毒素M1(AFM1)的检测。利用适配体和作为Cas12a/crRNA 的激活链,当AFM1存在时,适配体与靶标结合,则无法结合Cas12a/crRNA,此时Cas12a 没有活性,纳米金表面的ssDNA 引物和磷酸基团修饰的ssDNA 锁式探针能够在T4 连接酶和Phi29 DNA 聚合酶的作用下发生RCA 反应,生成大量的单链环绕在纳米金表面。加入对硝基苯酚后,体系呈现出绿色。若是没有AFM1,则适配体与Cas12a/crRNA 结合进而激活反式切割活性,能够将纳米金表面的ssDNA 引物和磷酸基团修饰的ssDNA 锁式探针切断,则无法发生后续的RCA 扩增,导致纳米金表面裸露。加入对硝基苯酚后,在纳米金的类过氧化物酶活性的催化下,对硝基苯酚被氧化成氨基苯酚,体系由绿色变为无色。通过颜色的变化实现AFM1 的超灵敏检测,最低检测限为0.05 ng/L。脂多糖(LPS)是一种内毒素,Wang 等[57]基于适配体、金属核酸酶和CRISPR/Cas12a 建立了CRISPR/Cas-纳米机器荧光传感器,用于LPS 的检测。将肼基标记ssDNA(ASI)和镁离子金属核酸酶的底物链(SS)修饰在磁珠(MB)表面,构建MB@SS/ASI复合物,醛基标记的ssDNA(ASII)中包含有金属核酸酶的酶链,ASII 能够通过C=N 键与ASI 相连,组装为MB@SS/ASI/ASII 纳米机器。随后ASII的酶链部分与SS 互补配对组装成完整的金属核酸酶,在镁离子的作用切断SS,释放出游离的单链靶标,ASII 则再次与新的SS 链互补并切割,往复循环至SS 全被切断。通过磁分离将游离的单链靶标分离,与Cas12a/crRNA 结合进而激活反式切割活性,荧光信号显著增强。在LPS 的检测体系中,ASII 中含有适配体互补序列,与适配体形成双链而封闭活性,当LPS 存在时,能够与适配体结合释放ASII,触发后续反应,荧光增强。如果没有LPS,则ASII 与适配体维持双链状态,进而无法触发后续反应,没有荧光信号。通过荧光信号强度的变化实现LPS 的检测分析,最低检测限为7.31 fg/mL。
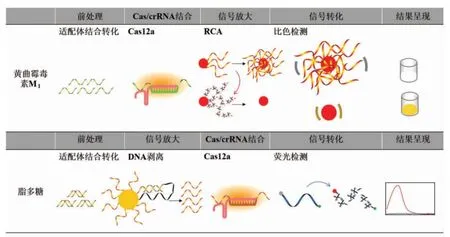
图3 生物毒素的CRISPR/Cas 核酸传感器检测流程示意图Fig.3 Schematic diagram of CRISPR/Cas nucleic acid biosensors detection process for biotoxin
3.3 金属离子的检测
食品安全风险检测中提及的重金属包含铅、汞、银、镉、铬等,目前的传感器快检技术主要是依基于金属离子对应的核酸酶开展研究,实现重金属离子向核酸信号的转化,同样,CRISPR/Cas 核酸传感器也采取相同的策略(图4)。Li 等[50]基于金属核酸酶和CRISPR/Cas12a 建立了CRISPR/Cas-荧光传感器,用于铅离子的检测。将铅离子金属核酸酶修饰在磁珠表面,并根据底物链设计crRNA,由于底物链互补且没有PAM 序列,导致其无法激活Cas12a 的活性,荧光信号不变。当铅离子存在时,切割底物链释放出游离的ssDNA,进而与Cas12a/crRNA 结合并激活反式切割活性,荧光信号增强。通过荧光的变化实现铅离子的检测,最低检测限为0.053 nmol/L。除重金属外,食品中钠离子作为核心营养素也备受关注,过多摄入也会影响人体健康。Xiong 等[58]基于金属核酸酶和CRISPR/Cas12a 建立了CRISPR/Cas-荧光传感器,用于钠离子的检测。将钠离子金属核酸酶底物链通过生物素-链霉亲和素作用进行固定,没有钠离子时,底物链与酶链组装为完整的金属核酸酶,无法激活Cas12a 的活性,荧光信号不变。当钠离子存在时,能够切割底物链释放出游离的ssDNA,进而与Cas12a/crRNA 结合并激活反式切割活性,荧光信号增强。通过荧光的变化实现钠离子的检测,最低检测限为0.1 nmol/L。
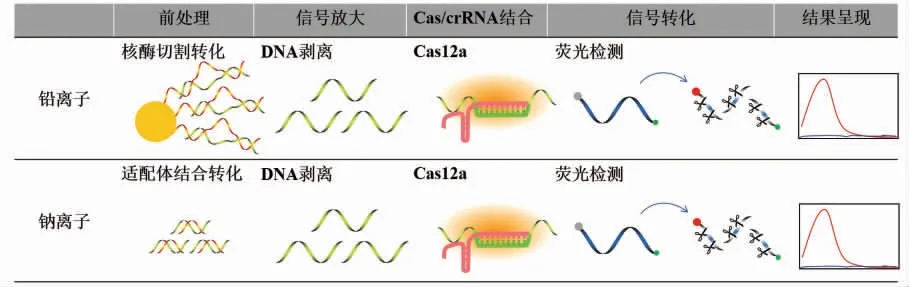
图4 金属离子的CRISPR/Cas 核酸传感器检测流程示意图Fig.4 Schematic diagram of CRISPR/Cas nucleic acid biosensors detection process for metal ions
3.4 生物技术食品的检测
生物技术食品一般是指基于生物技术得到的食品,目前大家比较熟识的主要是转基因食品和基因编辑食品。采用CRISPR/Cas 核酸传感器检测需要提取基因组、核酸扩增和Cas 蛋白切割3 个步骤(图5)。Wu 等[59]基于核酸扩增、CRISPR/Cas12a 和自主便携设备建立了CRISPR/Cas-便携式荧光传感器,用于转基因大豆的检测。通过基因组提取,针对35S 启动子和大豆内源基因(Lectin)进行双重LAMP 扩增,随后将反应管倒置,扩增产物分别与两个反应孔中的Cas12a/crRNA 结合,利用反式切割活性切割荧光淬灭探针,通过两个孔的荧光变化完成转基因大豆的检测,最低检测限为0.1%。Liu 等[60]基于核酸扩增和CRISPR/Cas12a建立了CRISPR/Cas-荧光传感器,用于转基因玉米的检测。通过基因组提取进行RPA 扩增,随后将扩增产物与Cas12a/crRNA 结合,在反式切割作用下通过荧光变化完成转基因玉米中NOS 终止子和35S 启动子的检测,最低检测限为10 拷贝。
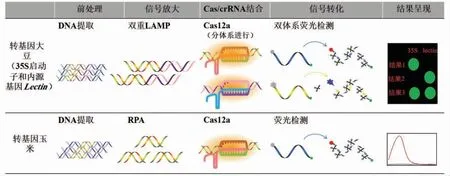
图5 生物技术食品的CRISPR/Cas 核酸传感器检测流程示意图Fig.5 Schematic diagram of CRISPR/Cas nucleic acid biosensors detection process for biotech foods
3.5 肉制品成分的检测
肉制品中的肉类掺假问题是全球热点,从我国的“挂羊头卖狗肉”到国外的“马肉事件”,以次充好、以假乱真的现象越来越常见。采用CRISPR/Cas 核酸传感器检测同样需要提取基因组、核酸扩增和Cas 蛋白切割3 个步骤(图6)。Liu 等[60]基于核酸扩增和CRISPR/Cas12a 建立了CRISPR/Cas-荧光传感器,用于肉制品中肉类成分的检测。通过基因组提取进行RPA 扩增,随后将扩增产物与Cas12a/crRNA 结合,在反式切割作用下通过荧光变化实现猪肉、牛肉和鸭肉的真实性检测,最低检测限分别为100 拷贝、10 拷贝和10 拷贝。

图6 肉制品的CRISPR/Cas 核酸传感器检测流程示意图Fig.6 Schematic diagram of CRISPR/Cas nucleic acid biosensors detection process for meat product
3.6 非法添加物的检测
一些食品安全事件是由于非法添加引起的,例如2008年的“三聚氰胺事件”,对中国奶业商业信誉造成了重创,就是因为生产者违法向奶粉中添加“非法添加物”三聚氰胺,导致众多婴幼儿出现肾结石甚至肾损伤。采用CRISPR/Cas 核酸传感器检测则需要将非法添加物转化成核酸进行分析(图7)。Qiao 等[61]基于适配体和CRISPR/Cas12a 建立了两个CRISPR/Cas-锁式荧光传感器,用于三聚氰胺的检测,检测准确性及灵敏度能够与HPLC相匹配。第1 个传感器中,利用一条互补链与适配体配对,当三聚氰胺与适配体结合后释放互补链,与Cas12a/crRNA 结合并激活反式切割活性,通过荧光强度的变化完成检测,最低检测限为38 nmol/L;第2 个传感器中,利用一条互补链与两条适配体配对,后续反应一致,最低检测限可达到0.24 μmol/L。
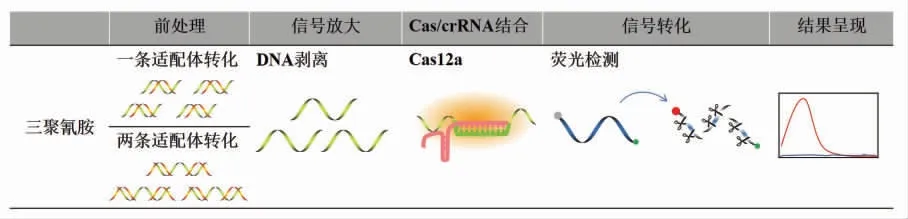
图7 非法添加物的CRISPR/Cas 核酸传感器检测流程示意图Fig.7 Schematic diagram of CRISPR/Cas nucleic acid biosensors detection process for illegal additives
4 展望
CRISPR/Cas 核酸传感器具有诸多优势,但在未来的发展中仍具挑战。第一,多重检测发展有局限,虽然能够通过不同的Cas 蛋白的选择性或者分离反应能够实现多重检测,但是方法的通用性不强。尤其是针对食品安全风险检测而言,能够实现单一体系下的多重检测更有研究意义,因此未来还应该在CRISPR/Cas 的多重分析上寻求新的突破,实现多重检测可行化、简便化。第二,核酸扩增技术虽能够显著放大原始信号,提升传感器的灵敏度,但是扩增过程中容易引起交叉污染造成结果假阳性。针对这一弊端虽然也有多种技术进行改进,但是却增加了操作上的繁琐,未来还需要进一步探索出更有效的方法。第三,结果呈现方面有比色、荧光、电信号等,其中液相的比色分析、部分荧光分析和固相的试纸条分析能够实现裸眼检测,能够应用在现场分析,但是电化学平台则推广受限,未来应该开发更多具有自主知识产权的小型便携设备,扩大基础科研走向现场应用。第四,食品安全检测应用方面,虽然涵盖了食源性致病菌、生物毒素等核酸和非核酸靶标,但是相关的研究并不多,并且大部分是以CRISPR/Cas12a 系统开展的研究,此外在农兽药残留检测等关键风险因子方面仍没有相关的研究,未来还需要进一步拓展CRISPR/Cas 的应用范畴。
猜你喜欢
北方牧业(2023年13期)2023-07-28 06:50:54
军民两用技术与产品(2021年10期)2021-03-16 06:05:10
无机化学学报(2020年7期)2020-07-20 02:06:44
三农资讯半月报(2020年8期)2020-05-13 14:26:35
世界农药(2019年3期)2019-09-10 07:04:10
中国病理生理杂志(2017年2期)2017-01-17 03:59:14
现代检验医学杂志(2016年3期)2016-11-15 01:59:28
光学精密工程(2016年4期)2016-11-07 09:04:48
三峡大学学报(自然科学版)(2016年6期)2016-04-16 05:02:56
肿瘤影像学(2015年3期)2015-12-09 02:38:45
