
UPLC-MS/MS法测定大鼠血浆甘草酸及其代谢物的浓度及药物动力学研究
2023-03-20 05:01:20张明玥沙丽娜刘晓航高晓黎
新疆医科大学学报 2023年1期
单 宇, 张明玥, 沙丽娜, 张 亮, 刘晓航, 高晓黎,4
(1新疆医科大学药学院, 乌鲁木齐 830017; 2新疆维吾尔自治区药品检验研究院, 乌鲁木齐 830054;3新疆第二医学院, 新疆 克拉玛依 834000; 4新疆及中亚特色医药资源教育部工程研究中心, 乌鲁木齐 830017)
甘草酸是中药甘草的主要活性成分,具有抗炎、免疫调节及抑制病毒增殖和对病毒灭活、促进肝细胞增殖作用[1-3]。甘草酸制剂广泛用于治疗各种病毒性肝炎[4-6]、药物性肝病[7-9]、肝硬化[10],湿疹[11]、银屑病[12]、系统性红斑狼疮[13-14]等。复方甘草酸苷片中主要活性成分甘草酸苷,由一分子甘草次酸和两分子葡萄糖醛酸组成,口服后甘草酸苷水解为甘草酸,其中大部分甘草酸被肠道菌群代谢为甘草次酸吸收入血,剩余的甘草酸在肠道吸收入血在肝脏中代谢为甘草次酸,甘草次酸的药理活性强于甘草酸[15-16]。体内甘草酸的含量受制剂、肠道菌群、肝脏代谢水平等因素影响,远低于甘草次酸[17-19]。目前体内甘草酸和甘草次酸的检测方法主要有HPLC-UV和LC-MS/MS等方法[20-23],但同时测定甘草酸及甘草次酸的分析方法较少。本实验探索建立更灵敏、高效的UPLC-MS/MS法同时检测大鼠血浆中甘草酸及甘草次酸的含量,现报道如下。
1 仪器与试药
1.1 仪器ACQUITY 型超高效液相色谱仪,XEVO-TQS micro型三重四级杆串联质谱联用仪 (美国Waters公司);高速台式低温离心机(英国Thermo Fisher Scientific公司);Milli-Q A10型纯水仪(密理博公司);MS105 型十万分之一分析天平(梅特勒-托利多公司)。
1.2 药品与试剂甘草酸(上海源叶生物科技有限公司,B20417,纯度≥98%)、甘草次酸(上海源叶生物科技有限公司,B20412,纯度≥98%)、格列喹酮(上海源叶生物科技有限公司,B20444,纯度≥98%),甲醇、乙腈(Thermo Fisher Scientific公司,LC-MS级),乙酸铵(CNW Technologies公司,HPLC级),复方甘草酸苷片(美能®)(Akiyama Jozai Co.,Ltd,17322)。
1.3 动物SPF级SD 大鼠雌雄对半,体重275±25 g,由新疆医科大学动物实验中心提供,实验动物许可证号为SYXK(新)2018-0003;饲养条件:普通级IVC饲养系统,温度 20~25℃,湿度40%~70%,光照12 h,自由进食与饮水。
2 方法与结果
2.1 色谱条件色谱柱:ACQUITY UPLC BEH C18(2.1 mm×150 mm,1.7 μm);流动相:5 mmoL/L乙酸铵(A)-乙腈(B),梯度洗脱(0~0.3 min:80%B,1.8~5 min:10%B,5.5~8 min:80%B),流速:0.3 mL/min;柱温:30℃;进样量:5 μL。
2.2 质谱条件离子源为电喷雾离子源(ESI),负离子检测模式;三重四极杆分析器;扫描方式为多反应离子监测(MRM)。脱溶剂温度:350℃;脱溶剂气流:800 L/Hr;待测物及内标检测参数见表1。用于定量的离子反应分别为甘草酸[M-H]-821.5/351.1、甘草次酸[M-H]-469.4/355.4、格列喹酮[M-H]-526.3/386.1。
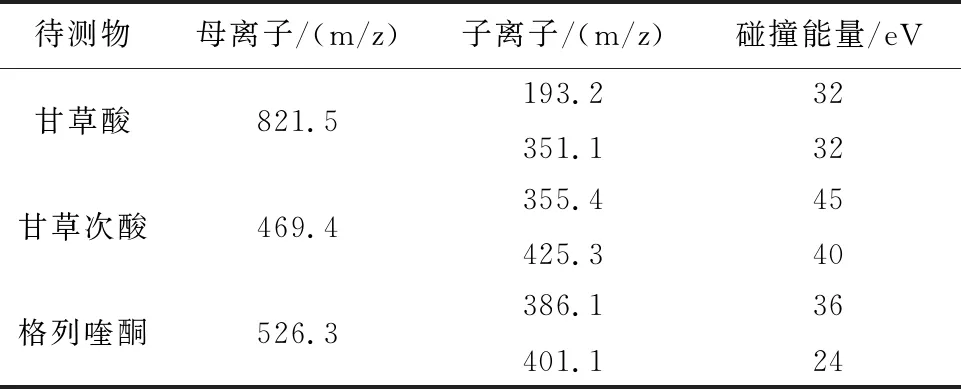
表1 各待测物母离子、子离子、驻留时间、锥孔电压及碰撞能量
2.3 溶液配制
2.3.1 对照品储备液制备 精密称取甘草酸对照品、甘草次酸对照品适量分别置于10 mL容量瓶中,甲醇溶解并稀释至刻度,制得浓度分别为200 μg/mL、4 000 μg/mL甘草酸和甘草次酸对照品储备液。精密称取内标格列喹酮适量置于100 mL容量瓶,甲醇溶解并稀释至刻度,摇匀制得150 ng/mL格列喹酮内标溶液,以上溶液均储存于4℃冰箱。
2.3.2 系列对照品溶液制备 从冰箱中取出各对照品储备液,放置室温后,用甲醇稀释成不同浓度的系列溶液,最后按其浓度级别混合均匀,得1~11号混合对照品系列质量浓度的溶液(甘草酸:0.1、0.2、0.5、1、3、6、9、12、18、24、29 μg/mL;甘草次酸0.4、9、28、67、90、112、140、168、196、224、269 μg/mL)。
2.4 血浆样品前处理取100 μL血浆样品于1.5 mL离心管中,加入内标溶液10 μL,涡旋2 min,加入1 000 μL甲醇沉淀蛋白,2 000 r/min涡旋混匀5 min,离心(4℃,14 000 r/min)10 min 得上清液1,取100 μL上清液1, 离心(4℃,14 000 r/min)10 min,得上清液2作为甘草次酸测试样品,进行UPLC-MS/MS分析。另取上清液850 μL,40℃水浴N2吹干,加90 μL甲醇复溶,3 000 r/min涡旋5 min,离心(4℃,14 000 r/min)10 min,得上清液3作为甘草酸测试样品进行UPLC-MS/MS分析。
2.5 方法学考察
2.5.1 专属性考察 分别取大鼠的空白血浆,配制空白样品、甘草酸与甘草次酸对照品血浆质量控制样品,以及大鼠灌胃复方甘草酸苷片后的样品,按“2.4”项下血浆样品前处理方法处理后进行UPLC-MS/MS分析。由图1可见,甘草酸、甘草次酸和内标的保留时间分别为3.14、4.96、4.31 min,甘草酸、甘草次酸血浆中基质与内源性物质无干扰,内标无干扰。表明本方法对甘草酸、甘草次酸专属性强。
2.5.2 标准曲线与定量下限 取1~11号混合对照品溶液10 μL,加入空白血浆90 μL,涡旋2 min,得混合对照品血浆,按“2.4”项下血浆样品前处理方法处理,按色谱与质谱条件进样测定,以甘草酸或甘草次酸浓度为横坐标,甘草酸或甘草次酸与内标峰面积比值为纵坐标,采用加权(W=1/X2)最小二乘法进行回归运算。甘草酸线性方程为:Y=0.545 051X+0.010 71(r=0.994 8),线性范围为:0.009 9~2.899 0 μg/mL,定量下限为0.009 0 μg/mL;甘草次酸线性方程为:Y=1.77 627X-0.004 328(r=0.998 3),线性范围为:0.040 0~26.890 0 μg/mL,定量下限为0.040 0 μg/mL。
2.5.3 准确度与精密度 按“2.2.3”方法,将储备液稀释得到12~14号混合对照品高、中、低浓度溶液(甘草酸:0.3、5、15 μg/mL;甘草次酸1.2、100、150 μg/mL)取12~14混合对照品高、中、低浓度溶液10 μL,加入空白血浆90 μL,涡旋2 min,得混合对照品血浆,平行配制 6 份,按“2.4”项下血浆样品前处理方法处理,按色谱与质谱条件进样测定,连续制备测定 3 批,按照当日标准曲线计算准确度,计算甘草酸苷与甘草次酸的准确度、日内、日间精密度。甘草酸苷与甘草次酸准确度在80%~120 %之内,日内精密度与日间精密度试验的RSD%均小于20%,准确度好、精密度高,符合分析方法的要求,见表2、3。
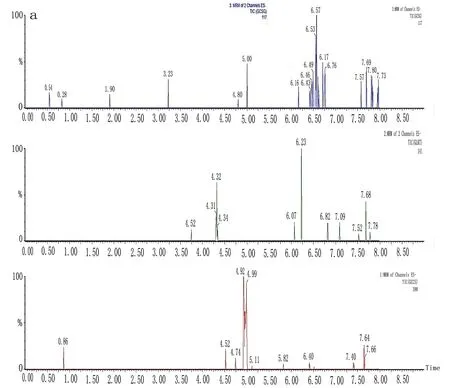
注:a为空白血浆;b为甘草酸与甘草次酸对照品血浆质量控制样品;c为大鼠灌胃复方甘草酸苷片后的样品。
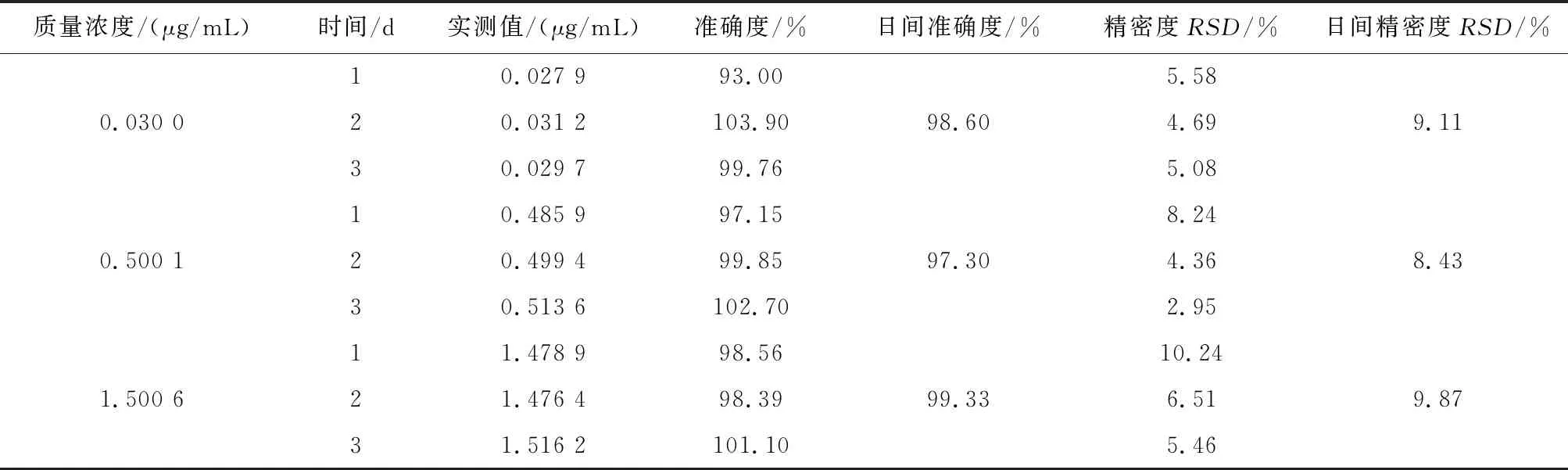
表2 血浆中甘草酸准确度、日内精密度、日间精密度(n=6)
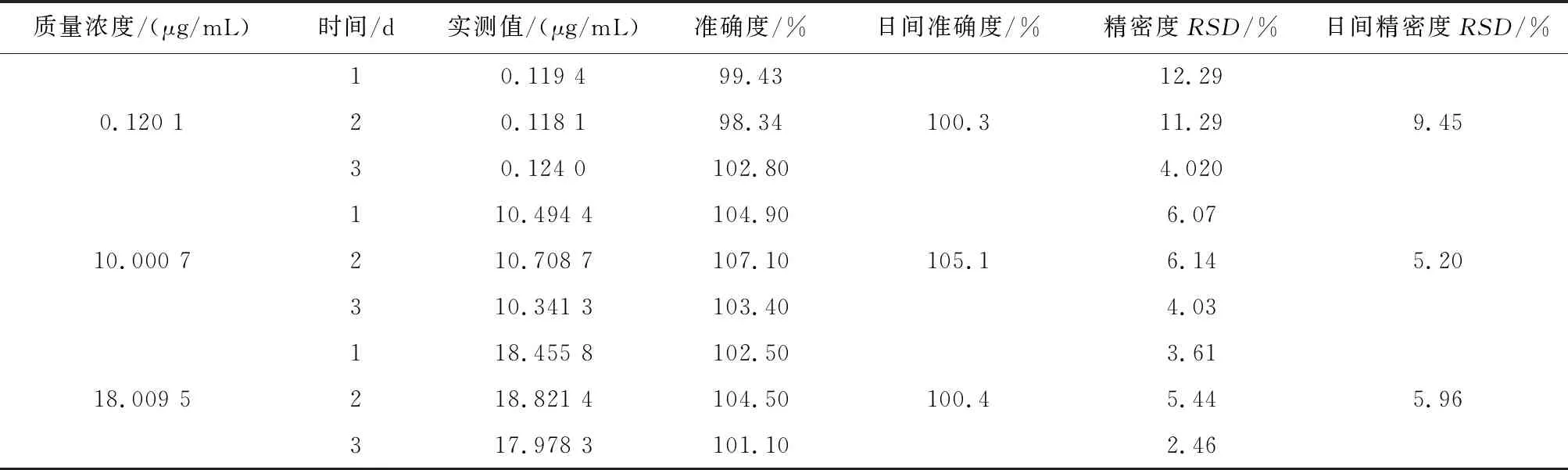
表3 血浆中甘草次酸准确度、日内精密度、日间精密度(n=6)
2.5.4 提取回收率及基质效应 按“2.5.3”项制备低、中、高混合对照品血浆,按“2.4” 项下方法处理后测定,该组峰面积记作A。另取空白血浆按“2.4”项下方法处理,取980 μL于EP管中,分别加入10 μL低、中、高三个浓度的甘草酸与甘草次酸对照品血浆质量控制样品的混合工作溶液与 10 μL格列喹酮内标溶液,涡旋震荡2 min,离心(4℃,14 000 r/min)10 min 得上清液1,取100 μL上清液1, 离心(4℃,14 000 r/min)10 min,得上清液2作为甘草次酸测试样品进行UPLC-MS/MS分析。另取上清液850 μL,40℃水浴N2吹干,加90 μL甲醇复溶,3 000 r/min涡旋 5 min,离心(4℃,14 000 r/min)10 min,得上清液3作为甘草酸测试样品进行 UPLC-MS/MS 分析,按色谱和质谱条件进样测定,该组峰面积记作 B。将与 B 组质量浓度相同的低、中、高3个浓度甘草酸与甘草次酸对照品混合溶液以及格列喹酮内标溶液分别按色谱和质谱条件进样测定,该组峰面积记作C。分别计算低、中、高血浆质量控制样品及内标的提取回收率(提取回收率=A/B×100%);基质效应:分别计算低、中、高血浆质量控制样品及内标的基质效应(基质效应=B/C×100%),得出分析物与内标的基质因子。甘草酸、甘草次酸的提取回收率,基质效应均在90%左右,表明方法的提取回收率较高且稳定,质谱响应离子增加或抑制效果不明显,符合对生物样品测定要求,结果见表4。
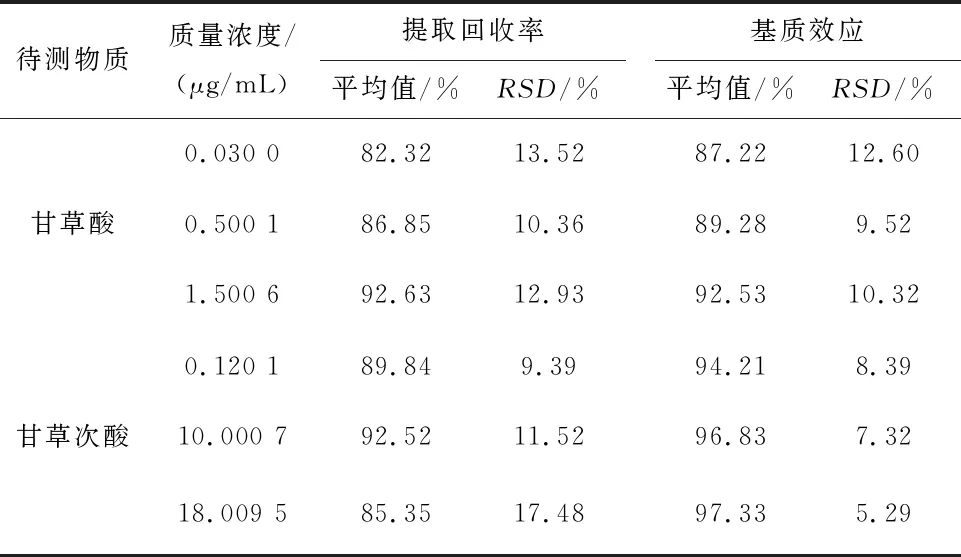
表4 血浆中甘草酸与甘草次酸提取回收率与基质效应(n=6)
2.5.5 稳定性试验 考察了血浆样品的短期稳定性和长期稳定性。短期稳定性包括血浆样品室温放置 4 h提取处理,进样器10℃放置24 h,及-20℃3次冻融循环后血浆样品的稳定性;长期稳定性考察-80℃冰箱放置 15 d血浆样品的稳定性。在上述各条件下,样品均保持稳定,结果见表5。
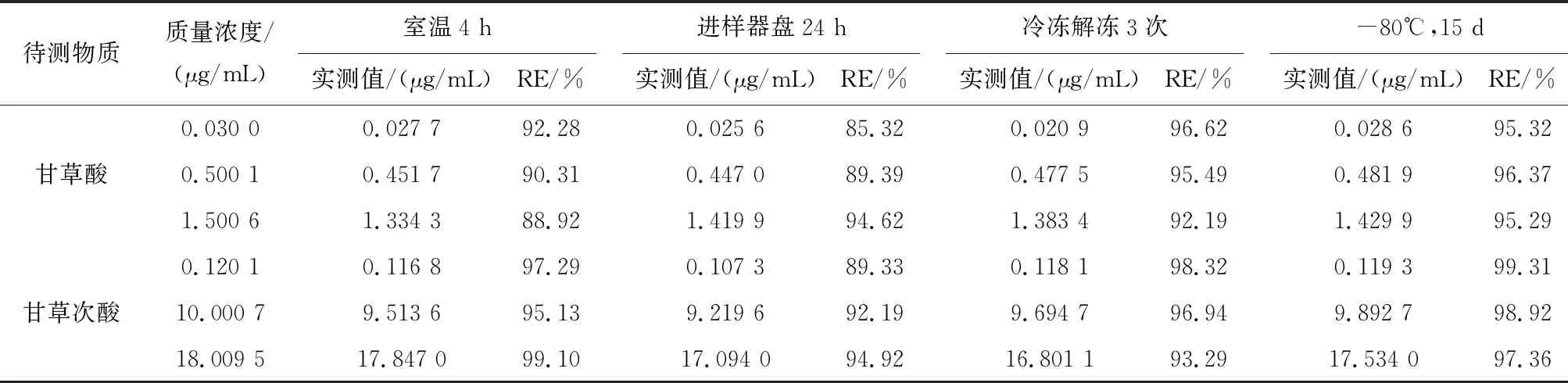
表5 血浆中甘草酸与甘草次酸样品的稳定性(n=6)
2.5.6 残留效应 定量上限样品后立即分析 3 针空白样品,并在 3 批标准曲线样品后重复操作,空白样品在甘草酸、甘草次酸、格列喹酮的保留时间均无响应,且在生物样品分析期间随行空白样品均无响应,此方法无残留效应,符合生物样品分析方法的要求。
3 药动学试验
3.1 试验方法SD大鼠12只,雌雄各半,给药前12 h禁食,自由饮水,动物在实验期间自由饮水,于给药后3 h自由进食标准SPF级鼠粮。大鼠按剂量6.75 mg/kg灌胃复方甘草酸苷片溶液,于0、0.5、1、1.5、2、3、5、7、8、9、10、12、24、50 h眼眶静脉丛采血,置于肝素化的EP管中,于4℃,5 000 r/min离心10 min,分离血浆,置于-80℃冰箱冷冻贮存备用,作为样品血浆,总采血量1.4 mL,并对大鼠及时补液。
3.2 试验结果采用Winnonlin软件,非房室模型统计分析其药动学参数,甘草酸、甘草次酸血药浓度-时间曲线分别如图2和图3所示,药动学参数结果如下:甘草酸Tmax为(1.83±0.41) h,Cmax为(0.42±0.04) μg/mL,AUC0-∞为(2.13±0.43) h·μg-1·mL-1,t1/2为(16.74±8.45) h。代谢产物甘草次酸Tmax为(7.5±1.64) h,Cmax为(3.58±0.65)μg/mL,AUC0-∞为(25.24±0.86) h·μg-1·mL-1,t1/2为(8.97±3.22) h。
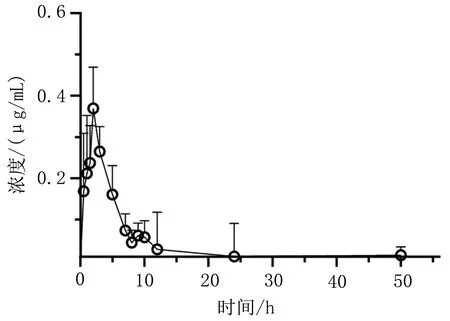
图2 甘草酸血药浓度-时间曲线(n=6)
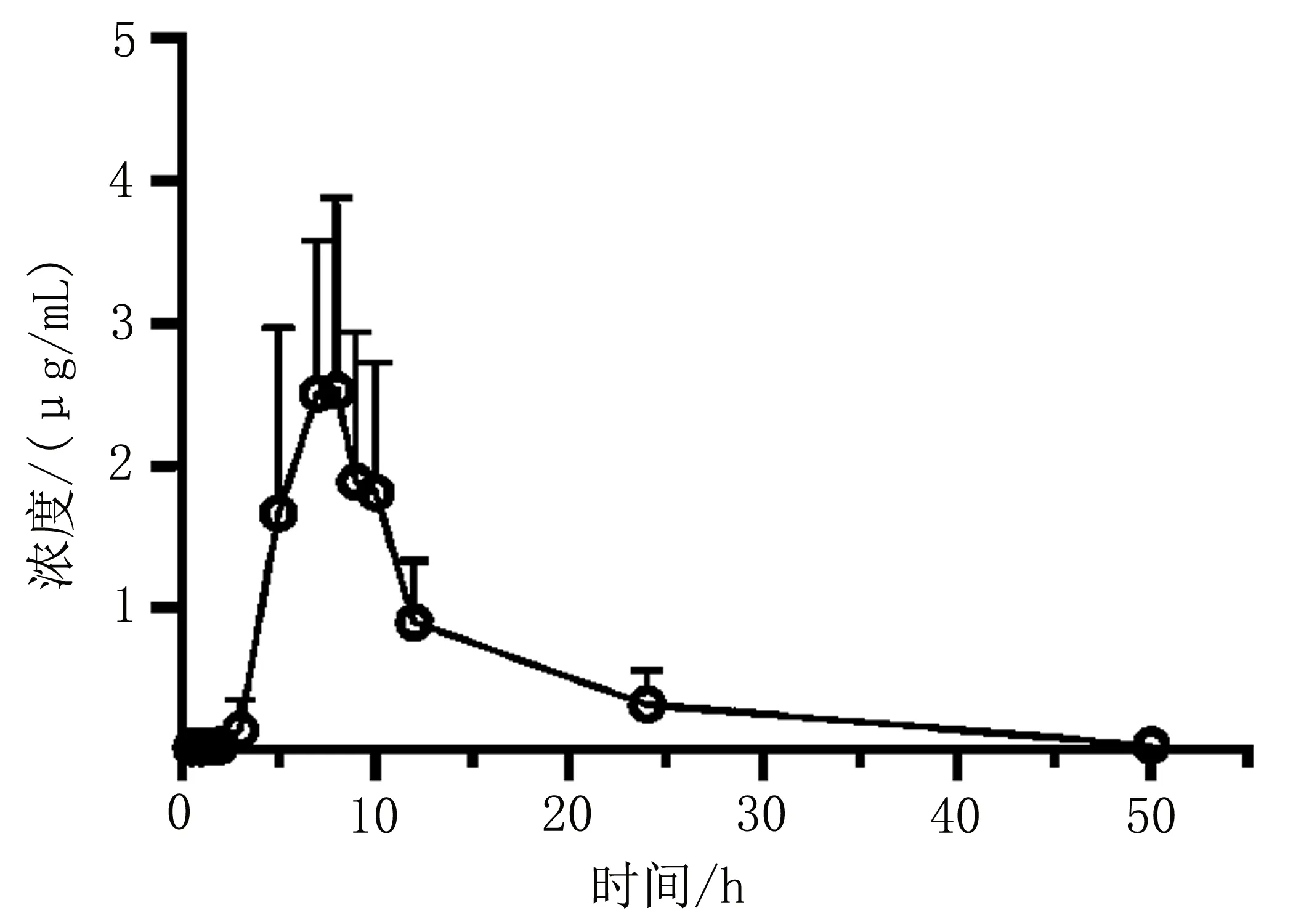
图3 甘草次酸血药浓度-时间曲线(n=6)
4 讨论
常见的血浆样品前处理方式为液液萃取法与沉淀蛋白法[23],根据甘草酸及甘草次酸的理化性质,本实验对血浆样品的前处理方法进行筛选,其中液液萃取法用乙酸乙酯提取后的血浆样品与待测物质存在干扰且存在乳化现象,提取回收率较低,故本实验采取沉淀蛋白法。前期实验对蛋白沉淀剂进行筛选,血浆在流动相混合溶液作为沉淀剂时,处理后可见混悬物,效果不好;乙腈作为蛋白沉淀剂时,处理后样品澄清透明,但每次测定分析后,色谱柱压力大幅度增加,易损伤色谱柱。将蛋白沉淀剂改为甲醇,进行沉淀剂体积考察后,发现最终确定的血浆前处理方法待测物与内源性物质无干扰,色谱图峰型较好,故采用甲醇沉淀法作为本实验的前处理方法。
甘草酸苷制剂口服后,甘草酸苷经胃酸水解或经肝中β-葡萄糖醛酸苷酶水解为甘草次酸,甘草次酸在肝肠循环中经肠内菌作用部分生成 3-表-甘草次酸及少量的 3 -脱氢甘草次酸[19],最终代谢为甘草次酸。本实验中部分大鼠甘草次酸药时曲线出现双峰,证明甘草次酸进入体内后存在肠肝循环,提示该品种的吸收存在较大的个体间差异,人体生物等效性的研究需考虑增大试验样本量,得到更可靠的结果。
本试验建立了可同时准确地检测血浆中甘草酸及甘草次酸含量的UPLC-MS/MS方法,线性范围广,灵敏度高,检测时间短。该分析方法可为甘草酸苷制剂人体生物等效性研究、甘草酸与甘草次酸的血药浓度检测提供参考。
猜你喜欢
酿酒科技(2023年10期)2023-11-23 11:09:42
食品安全导刊(2019年27期)2019-12-09 07:34:16
名城绘(2019年4期)2019-10-21 05:09:13
中成药(2018年2期)2018-05-09 07:20:08
中成药(2017年3期)2017-05-17 06:08:48
中国卫生标准管理(2015年4期)2016-01-14 05:16:45
山东医药(2015年16期)2016-01-12 00:40:04
西南医科大学学报(2016年4期)2016-01-03 01:26:29
饲料博览(2015年12期)2015-04-04 04:28:36
中国当代医药(2015年33期)2015-03-01 02:09:17
