
八味通里颗粒的质量标准研究
2023-03-15 23:34:16赵昱玮司学玲张瀚水龚本义南敏伦赫玉芳赵红菲
中国医药科学 2023年3期
赵昱玮 司学玲 任 辉 张瀚水 龚本义 南敏伦 赫玉芳 赵红菲
1.长春中医药大学附属医院制剂中心,吉林长春 130117;2.修正药业集团长春高新制药有限公司质量管理部,吉林长春 130012;3.吉林大学中日联谊医院普外科,吉林长春 130033;4.长春工业大学化学与生命科学学院,吉林长春 130021;5.吉林省中医药科学院植物化学研究所,吉林长春 130012;6.长春中医药大学健康管理学院,吉林长春 130117
八味通里颗粒由大黄、芒硝、炒莱菔子、厚朴、枳壳、槟榔、地黄、枳实组成。取自汉代《伤寒论》“大承气汤”加减而成。具有通腑泄热、滋阴润燥、行气活血的功效。用于阳明腑实所致大便秘结不通、脘腹胀满、疼痛拒按、高热神昏、谵语、口干舌燥,舌红苔黄腻,脉滑数属。可用于术后肠梗阻、单纯性肠梗阻、粘连性肠梗阻、急性胆囊炎、急性胰腺炎、急性阑尾炎等见上述侯证候者。方中大黄[1-2]苦寒泻热,祛瘀通便,荡涤肠胃邪热积滞,消除致病之因,用为君药。芒硝[3-4]咸苦而寒,泻热通便,润燥软坚,协大黄则峻下热结,用为臣药。积滞内阻,腑气不通,内结之积滞恐难速下,重用厚朴、枳壳、炒莱菔子、枳实、槟榔行气宽中,消胀除满,燥屎热结伤阴故使以生地滋阴润燥养血[5-14]。共成通腑泄热、滋阴润燥,行气活血之功效。
为了更好地控制该制剂的质量,本研究对八味通里颗粒中的炒莱菔子、大黄、槟榔进行薄层色谱法(thin-layer chromatography,TLC)鉴别,并通过高效液相色谱法(high performance liquid chromatography,HPLC)测定枳壳和枳实中新橙皮苷的含量。
1 仪器与材料
1.1 仪器
1260型高效液相色谱仪(Agilent);KQ2200DE型超声波清洗器(昆山舒美);QUINTIX125D-1CN型电子天平(德国Sartorius公司)。
1.2 试药
芥子碱硫氰酸盐、大黄素、大黄酚、新橙皮苷对照品(批号分别为111702-202006、110756-201913、110796-201922、111857-201804,纯度分别为99.0%、96.0%、99.4%、99.4%,中检院)。乙腈(色谱纯,Sigma),薄层板为青岛海洋化工厂分厂生产,其他试剂均为分析纯(AR)。八味通里颗粒(批号:20210901、20210902、20210903,吉林大学中日联谊医院)。
2 试验方法
2.1 薄层鉴别
2.1.1 炒莱菔子 称量研细后的八味通里颗粒3 g于具塞锥形瓶中,加25 ml无水乙醇,在50℃条件下超声40 min,置冷水中冷却后滤过,回收乙醇提取液,残渣用乙酸乙酯浸泡2次,10 min/次、每次20 ml,分离并弃去乙酸乙酯液,残渣挥干,加1 ml甲醇溶解,作为供试品溶液。莱菔子对照药材,磨成细粉,称取0.5 g,加水20 ml,按照供试品制备方法制成对照药材溶液。精密称取芥子碱硫氰酸盐对照品适量,加甲醇制成1 mg/ml的溶液,作为对照品溶液。照《中国药典》2020年版四部通则0502 TLC试验,吸取上述三种溶液各3~6 μ1,分别点于同一羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以乙酸乙酯-甲酸-水(20∶4∶6)放置分层的上层溶液为展开剂,于上行展开缸中展开后,取出,晾干,放置在三用紫外分析仪上在365 nm下观察。供试品色谱中,在与对照药材及对照品相应的位置上,显相同颜色的荧光斑点。见图1。
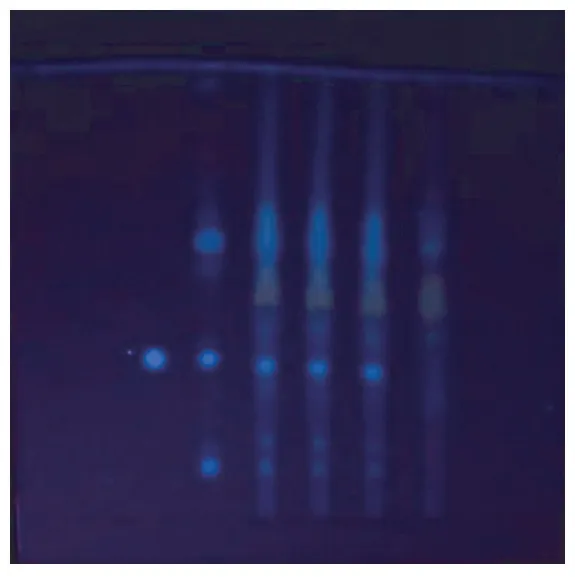
图1 炒莱菔子薄层色谱图
2.1.2 大黄 取八味通里颗粒3 g,研成细粉,加入25 ml甲醇,在50℃条件下超声30 min,放置至室温,滤过,滤液回收溶剂至干,加去离子水10 ml、浓盐酸1 ml,100℃加热回流水解30 min,冷却后,用三氯甲烷提取水解液2次,每次10 ml,合并两次三氯甲烷液,挥干溶剂,残渣加1 ml甲醇溶解,作为供试品溶液。另取大黄对照药材细粉0.1 g,按照供试品的制备方法制成对照药材溶液。再取大黄素、大黄酚适量,加三氯甲烷制成每1 ml分别含0.5 mg的溶液,为对照品溶液。照《中国药典》2020年版四部通则0502薄层色谱法试验,吸取上述三种溶液各1~3 μ1,点于同一羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以石油醚(60℃~90℃)-甲酸乙酯-甲酸(30∶10∶2)放置的上层溶液为展开剂,于上行展开缸中展开后,取出,自然晾干,放置在三用紫外分析仪上在365 nm下观察。供试品色谱中,在与对照药材及对照品色谱相应的位置上,显相同颜色的荧光斑点。见图2。
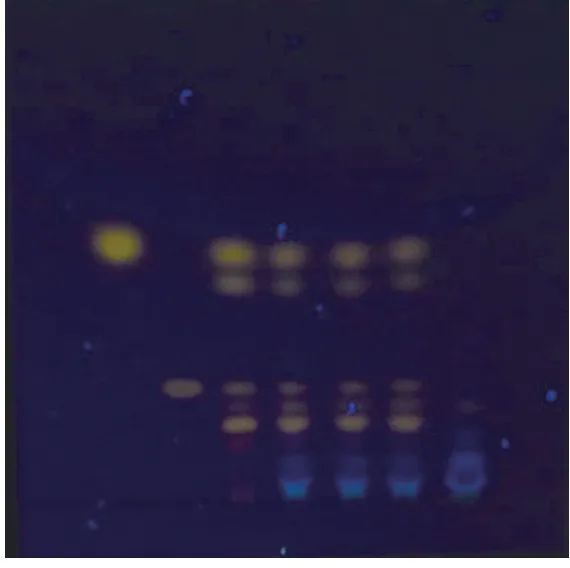
图2 大黄薄层色谱图
2.1.3 槟榔 取八味通里颗粒10 g,研成细粉,先加入2 ml浓氨试液,搅拌使粉末润湿、再加入40 ml三氯甲烷,摇匀,放置30 min,超声30 min,分取三氯甲烷液,用盐酸(1→5)溶液30 ml,振摇提取,取盐酸水层,用氨试液调节pH=9,再用三氯甲烷振摇提取2次,每次30 ml,合并三氯甲烷液,挥干,残渣加1 ml甲醇搅拌使溶解,为供试品溶液。另取槟榔对照药材粉末l g,按照上述供试品制备方法制成对照药材溶液。照《中国药典》2020年版四部通则0502薄层色谱法试验,吸取上述两种溶液各5~10 μl,点于同一羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以环己烷-乙酸乙酯-浓氨试液(15∶15∶0.2)为展开剂,置氨蒸气预饱和的展开缸内,上行展开都,取出,自然晾干,置放置碘的密闭空间中显色。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。见图3。

图3 槟榔薄层色谱图
2.2 新橙皮苷含量测定
2.2.1 色谱条件 分析柱为ACE-C(184.6 mm×250 mm,5 μm),以乙腈-0.25%磷酸溶液(19∶81)为流动相,检测波长283 nm,流速0.9 ml/min,柱温25℃,进样量10 μl。见图4。
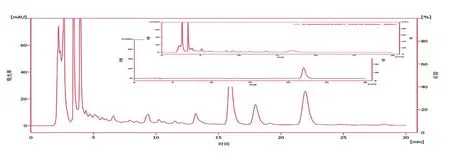
图4 HPLC色谱图
2.2.2 八味通里颗粒供试品溶液的制备 取八味通里颗粒,用研钵研成细粉,取约1 g,精密称重,置具塞锥形瓶中,加甲醇溶液25 ml,密塞,称定重量,超声处理(功率100 W,频率40 kHz)60 min,放置至室温,称重,加甲醇溶液补量,振荡均匀,滤过(0.2 μm),取续滤液,即得。
2.2.3 对照品溶液制备 精密称取新橙皮苷对照品适量,加甲醇超声溶解,制成含新橙皮苷质量浓度约为105 μg/ml的溶液。
2.2.4 线性关系考察 取对照品溶液各2、4、8、12、16、20 μl,分别利用液相色谱仪进行测定,横坐标(X)为新橙皮苷的进样量(μg),纵坐标(Y)为新橙皮苷的峰面积(A),绘制标准曲线,线性方程为Y=1496.2X-31.92,r=0.9998,结果表明,新橙皮苷含量在0.21~2.10 μg范围内线性良好。
2.2.5 精密度试验 取八味通里颗粒同一供试品溶液(20210902),照“2.2.1”色谱条件连续测定6次,新橙皮苷峰面积的RSD为0.65%(n=6),说明仪器精密度良好。
2.2.6 重复性试验 取八味通里颗粒(20210902),照“2.2.2”制备6份供重复性测定用的供试品溶液,照“2.2.1”色谱条件进行测定,新橙皮苷含量的平均值为1.61 mg/g,RSD为1.15%(n=6),说明该方法重复性良好。
2.2.7 稳定性试验 取八味通里颗粒(20210902)同一供试品溶液,于0、2、4、8、10、12、24 h各进行测定,新橙皮苷峰面积RSD为1.48%,说明样品在24 h内稳定性良好。
2.2.8 加样回收率考察 取八味通里颗粒(20210902)6份,每份约0.5 g,精密称定,分别精密加入浓度为0.8220 mg/ml新橙皮苷对照品1 ml,制备供试品溶液,按色谱条件测定,实验结果如表1。计算得到新橙皮苷的平均回收率为99.91%,RSD为0.43%。

表1 加样回收率实验结果
2.2.9 含量测定 取20210901、20210902、20210903批次八味通里颗粒样品,按照“2.2.2”下方法制备供试品并进行检测,3批颗粒新橙皮苷含量分别为1.58、1.61、1.60 mg/g,平均含量为1.60 mg/g。按照新橙皮苷不低于80%计,八味通里颗粒中新橙皮苷的含量应不低于1.20 mg/g。
3 讨论
3.1 薄层鉴别
对八味通里颗粒中各药味进行了摸索,最终建立了炒莱菔子、大黄、槟榔的薄层鉴别方法。参考《中国药典》2020年版一部及参考文献[15-20]中的薄层鉴别方法,对八味通里颗粒中炒莱菔子、大黄、槟榔鉴别方法进行优化,确定了最优的薄层色谱条件。
3.2 含量测定
①分别考察了甲醇-磷酸溶液、乙腈-磷酸溶液流动相系统[21-24],考察了ACE-C18、Agilent ODS-C18、Hypersil ODS-C18色谱柱,考察了不同的检测波长、柱温的影响,最终确定分析柱为ACE-C18(4.6 mm×250 mm,5 μm),以乙腈-0.25%磷酸溶液(19∶81)为流动相,检测波长283 nm,流速0.9 ml/min,柱温25℃,进样量10μl的色谱条件。②以提取的新橙皮苷含量为评价指标,对提取的溶剂(分别设定甲醇和乙醇)、提取溶剂用量(25、50 ml)、提取的方式(超声提取、加热回流提取)、提取时间(30、60、90 min)进行考察,最终确定的提取方法为甲醇25 ml,超声处里60 min。最终建立的HPLC测定八味通里颗粒中新橙皮苷含量专属性强、重复性好,结果可靠。
本研究建立了八味通里颗粒中炒莱菔子、大黄、槟榔的定性鉴别方法和新橙皮苷的HPLC含量测定方法,方法简便操作性强,可用于八味通里颗粒的质量控制提供依据。
猜你喜欢
中国民间疗法(2021年17期)2021-11-04 08:39:38
中国民间疗法(2021年7期)2021-07-22 06:44:10
中成药(2021年5期)2021-07-21 08:38:56
家庭医学(下半月)(2020年4期)2020-05-30 12:42:48
益寿宝典(2017年2期)2017-02-26 21:27:52
中成药(2016年4期)2016-05-17 06:07:58
吉林大学学报(医学版)(2015年3期)2015-12-17 07:47:51
妇女生活(2015年7期)2015-07-20 05:42:36
妇女生活(2015年7期)2015-07-20 05:42:36
中国当代医药(2015年24期)2015-03-01 02:06:02
