
结核分枝杆菌1-脱氧-D-木酮糖-5-磷酸还原异构酶抑制剂的3D-QSAR研究及优化设计
2023-03-14 03:51:58谢双龙林治华
重庆理工大学学报(自然科学) 2023年2期
谢 稳,谢双龙,余 娜,林治华
(1.重庆理工大学 药学与生物工程学院, 重庆 400054;2.重庆市公共卫生医疗救治中心药学部, 重庆 400036)
0 引言
结核病(Tuberculosis,TB)是由感染结核分枝杆菌(Mycobacterium tuberculosis,Mtb)所导致的世界性传染病之一,多发病于肺部,也可感染其他器官和组织,具有高发病率和高死亡率。根据世界卫生组织WHO出具的全球结核病报告2020年版的数据,在2019年全球约有1 000万人感染结核,140万人死于结核(其中有20万死亡患者为HIV阳性)[1]。目前,WHO推荐的药物敏感性结核病的治疗方案为四药联合,即患者使用抗结核一线药物异烟肼、利福平、吡嗪酰胺、乙胺丁醇强化治疗2个月后,再用异烟肼、利福平继续规范治疗4个月[2]。但随着多药耐药和广泛耐药结核病的增多,以及HIV患者感染结核病例更加普遍,寻求新的化学治疗手段成为了抗结核病研究的重点。
磷酸甲基赤藓糖醇(methylerythritolphosphate,MEP)途径是结核分枝杆菌用于生物合成异戊烯基焦磷酸和其异构体二甲基烯丙基焦磷酸的重要环节,后两者是异戊二烯的通用五碳前体。异戊二烯在细菌细胞壁成分的生物合成中起着关键作用,因此,对结核分枝杆菌和一些其他致病细菌的生存至关重要[3]。在人类体内,磷酸甲基赤藓糖醇途径则完全不存在,取而代之的是由甲羟戊酸途径合成人体所需的异戊二烯,因此,磷酸甲基赤藓糖醇途径体现出的系统选择性以及对细菌生存的重要性,使参与其中的代谢酶成为新型抗结核药物开发的潜在靶标[4]。1-脱氧-D-木酮糖-5-磷酸还原异构酶(1-deoxy-D-xylulose-5-phosphate reductoisomerase,DXR)是磷酸甲基赤藓糖醇途径中的第二个酶,催化第二个限速反应,在氧化还原辅助因子NADPH的协助和二价金属阳离子的存在下,催化1-脱氧-D-木酮糖-5-磷酸盐(1-deoxy-D-xylulose-5-phosphate,DXP)形成磷酸甲基赤藓糖醇[5]。膦胺霉素(Fosmidomycin)为从薰衣草链霉菌属中分离出的天然产物[6],对结核分枝杆菌1-脱氧-D-木酮糖-5-磷酸还原异构酶(mtDXR)表现出强大的体外抑制活性,IC50值为80 nM,能取代DXP和DXR、NADPH、Mn2+形成四元共晶结构,从而抑制催化反应进程[7]。然而,因为结核分枝杆菌的细胞壁成分中富含霉菌酸,高极性的膦胺霉素无法穿透其脂质细胞壁,从而无法产生整体细菌抑制活性[8]。因此,研究者们对膦胺霉素的化学结构进行了大量改造尝试,包括但不限于合成磷酸酯前药、修饰异羟肟酸基团以及主链改造等,合成了一系列膦胺霉素衍生物,以期提高其疏水性能和整体抑菌活性[9-12]。
本文在前人研究的基础上,收集了35个已证实对mtDXR有体外抑制活性的膦胺霉素衍生物,用比较分子场分析方法(comparative molecular field analysis,CoMFA)和比较分子相似性指数分析方法(comparative molecular similarity indices analysis,CoMSIA),建立了可靠的3D-QSAR模型,剖析其立体场、静电场和氢键受体场的三维等势图,再结合分子对接得到的小分子配体和蛋白靶点的作用模式,设计出14个新型的膦胺霉素类小分子化合物,并确定了有望作为先导化合物展开进一步研究的新型小分子27m,为全新mtDXR抑制剂的开发提供了理论参考。
1 研究方法
1.1 数据来源
本文研究的35个膦胺霉素衍生物均来源于文献[13-15],在其中随机挑选7个化合物作为测试集(前面标有符号“*”),剩下的28个化合物作为训练集。训练集用于建立3D-QSAR模型,而测试集用于验证基于训练集数据所建模型的预测能力。原文献中用IC50值表示化合物,对mtDXR的体外抑制活性,为了更好地输入和计算研究数据,本文中换算成其负对数(-log IC50)作为因变量生物活性的单位,即pIC50值,如表1所示。膦胺霉素衍生物的分子结构见图1。
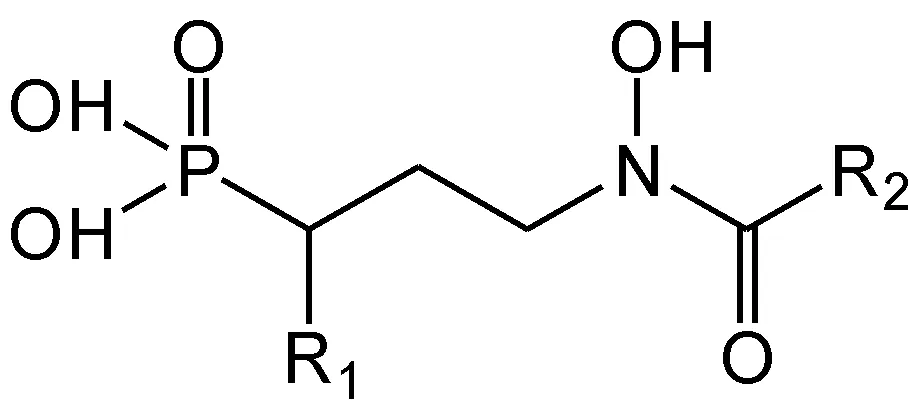
图1 膦胺霉素衍生物的分子结构示意图
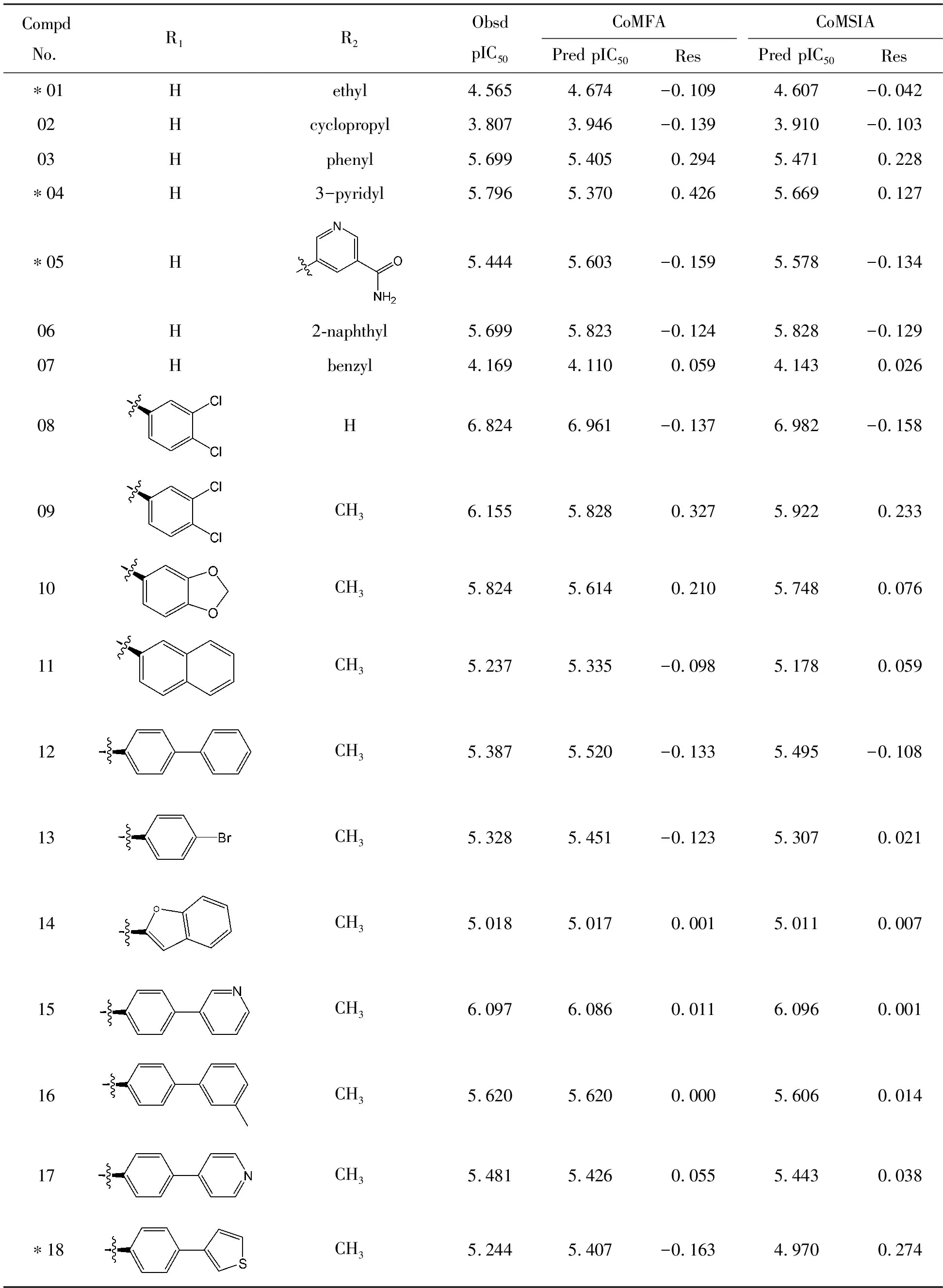
表1 膦胺霉素衍生物的实测活性与3D-QSAR模型预测活性值

续表(表1)
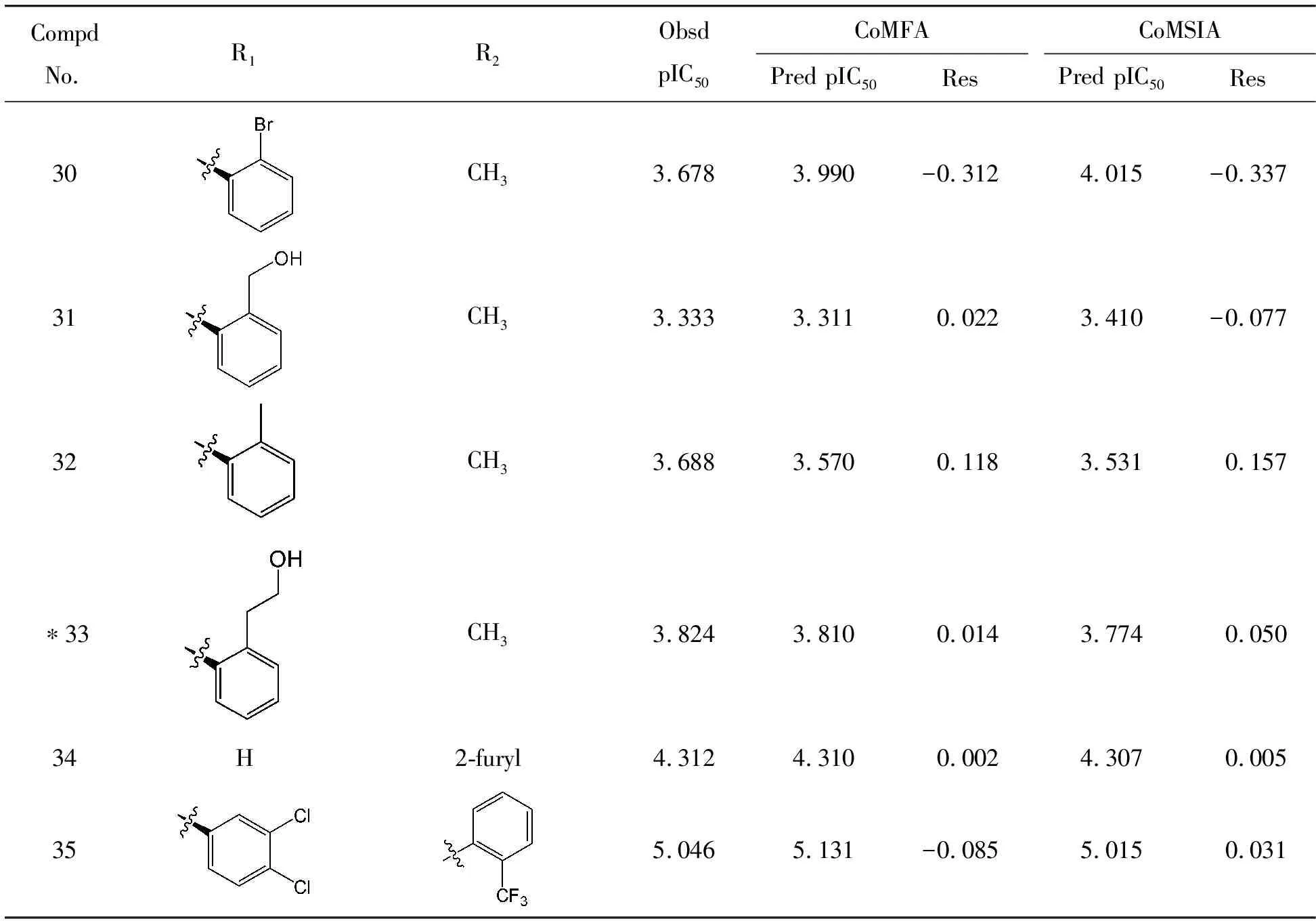
续表(表1)
1.2 分子结构优化和叠合
本文的35个膦胺霉素衍生物的三维分子结构均在软件SYBYL-X 2.1(来源:重庆理工大学)上搭建,加氢、命名,先用遗传算法GA conformational search筛选化合物分子的低能构象,然后给每个化合物分子赋予Tripos力场和Gasteiger-Huckel电场,利用Powell方法对化合物分子进行能量优化,最大迭代次数设置为 2 000,终点梯度设置为0.005 kcal/mol,其余参数为默认值。以展现出最高抑制活性的化合物27号作为叠合模板,将结构优化后的化合物构象基于公共骨架进行分子叠合,27号化合物的结构见图2(加粗部分为此次叠合的公共骨架)。基于公共骨架的分子叠合结果详见图3。

图2 化合物27的分子结构示意图

图3 基于公共骨架的分子叠合示意图
1.3 3D-QSAR模型的建立
在SYBYL-X 2.1的QSAR模块,运用经典的CoMFA和CoMSIA方法构建叠合化合物的3D-QSAR模型。比较分子场分析法(CoMFA)是Cramer等于1988年创建的三维定量构效关系研究方法[16],该方法认为,在分子水平上,药物分子周围立体场和静电场与受体之间的非共价键相互作用是其生物活性的主要来源,将叠合好的分子放置于一个三维网格中,使用一个sp3杂化、带+1价电荷的碳原子为探针,计算格点上探针与药物分子的相互作用能,以确定各种分子场的空间分布[17]。而比较分子相似性指数分析法(CoMSIA)于1994年由Klebe等提出[18],该方法与CoMFA方法最大的区别是在立体场和静电场以外,又定义了疏水场、氢键受体和氢键供体场这3种分子场,且分子场的计算采用了与距离相关的高斯函数,有效避免了CoMFA方法中由于采用Coulomb和Lennard-Jones势函数所引起的缺陷,且不再需要定义能量截断值。因作为自变量的分子场数值数目远大于作为因变量的生物活性数值,故采用偏最小二乘法(partial least squares,PLS)进行回归分析。首先使用留一法(leave-one-out,LOO)进行交互检验,得到最佳主成分值n和交互检验系数q2,普遍认为q2>0.5时,模型才有预测能力;然后再以得出的最佳主成分值进行非交互检验分析,得到相关系数r2,标准误差SEE和F检验值,可靠的F检验值理论上应大于100(F>100);再者应用建好的3D-QSAR模型对测试集和训练集的化合物进行活性预测,分析化合物的实测活性值与模型预测活性值之间的相关性;最后利用三维等势图(contour maps)显示QSAR方程,研究化合物公共骨架结构上的基团取代与整体生物活性之间的关系,从而在此基础上进行膦胺霉素衍生物分子结构的优化和改造,以设计得到预测活性更高的新型化合物。
1.4 分子对接分析
分子对接主要用于展示小分子配体和蛋白受体之间的相互作用,计算并预测其结合模式和亲和力,继而进行作用机理解释或先导化合物虚拟筛选,是基于靶点结构的药物设计的一种重要方法[19]。本文研究的靶点蛋白mtDXR的晶体结构下载于蛋白质结构数据库PDB(www.rcsb.org,code:2Y1D),使用SYBYL-X 2.1的蛋白准备模块,先对靶点蛋白结构进行计算分析,再在分析结果的基础上进行加氢、加电荷、修复侧链等处理,移去原配体小分子以便形成对接活性口袋,只保留A链氨基酸残基,移去所有水分子,修改His248的质子化状态,以便质子化的氮能朝向小分子抑制剂,其余参数保留为默认值[15]。使用Surflex-Dock Geom模式将膦胺霉素类小分子化合物对接到处理后的靶点蛋白的活性口袋中,分析小分子配体与活性位点关键氨基酸残基的非键作用,与3D-QSAR模型的结果互相验证,共同指导化合物分子结构的改造。
2 结果与讨论
2.1 3D-QSAR建模结果
以训练集28个膦胺霉素衍生物构建的CoMFA和CoMSIA模型的最佳统计学结果如表2所示。使用CoMSIA方法建模的过程中,可考虑各种不同分子场的组合以得到最佳模型,最后列出按照q2值降序排列条件下的前5名分子场组合的统计结果,见表3。立体场和静电场组合(S+E)得到的CoMFA模型,其最佳主成分值n=7,交互检验系数q2=0.601,大于0.5,代表此模型有较好的稳定性与预测能力,此外,较大的相关系数r2(0.979)和F值(131.383)以及偏小的标准误差SEE(0.159),也说明此模型有较强的拟合能力和统计学意义。就各种分子场组合的q2、F值和标准误差综合考虑,CoMSIA模型的最佳分子场组合为立体场、静电场和氢键受体场(S+E+A),其中立体场和静电场的贡献值分别为0.372%和0.564%,说明小分子抑制剂的活性主要与其中基团的立体效应和电荷分布相关,这与CoMFA模型的结果基本一致。最佳CoMSIA模型的PLS计算结果(见表2),也预示其有较好的预测能力。
为了对以上CoMFA和CoMSIA模型的预测能力进行验证,用模型先后预测了28个训练集化合物和7个测试集化合物的活性值(pIC50),然后用实测活性值与预测活性值(见表1)进行线性回归分析,从图4可以看出,绝大部分的化合物的活性数据都分布在趋势线附近,显示了预测活性值和实测活性值有着较高的拟合度,两大模型也都具有良好的预测能力。

表2 CoMFA和CoMSIA模型的最佳统计结果
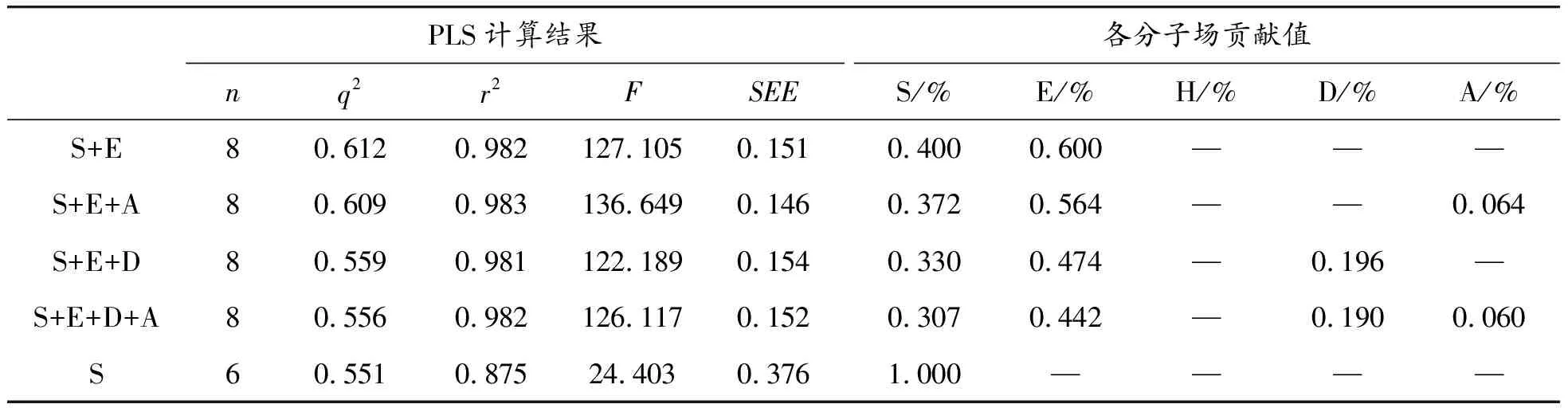
表3 按照q2值降序排列的前5名分子场组合下的CoMSIA模型统计结果

图4 训练集和测试集的预测活性值与实测活性值之间的相关性曲线
2.2 三维等势图结果及分析
CoMFA和CoMSIA模型的三维等势图以实测活性最高的27号分子为模板来展示。从统计结果可以看到,在CoMFA模型中,以立体场(0.602%)的贡献值为主,在CoMSIA模型中,以立体场(0.372%)和静电场(0.564%)的贡献值为主,而在后续的分子改造实践中,发现氢键受体在相关位置的引入也对提高预测活性有影响,因此,以此4个分子力场的三维等势图为基础来分析化合物分子结构上可进行修饰和基团改造的区域,如图5所示。
图5(a)是CoMFA模型立体场的三维等势图,绿色色块表示在此部位应加入大体积基团,而黄色色块表示此处的取代基团体积越小越有利。异羟肟酸部分的取代苯环的间位和对位有大面积的绿色色块,说明在此两处以大体积基团取代可以增加化合物活性,而邻位的黄色色块则说明小体积基团取代或不取代可以增加化合物活性,如化合物27(pIC50=6.854)和28(pIC50=5.921)的活性就大于化合物26(pIC50=4.886)。Cα部位的二氯苯环取代基上也有大面积的绿色色块,说明在此处增加立体位阻以较大体积的基团取代能增强化合物活性,如化合物16(pIC50=5.620)的活性就大于化合物12(pIC50=5.387)。

图5 27号模板分子的CoMFA和CoMSIA三维等势图
图5(b)是CoMSIA模型立体场的三维等势图,可以发现其分布与CoMFA模型基本一致,异羟肟酸部分的取代苯环的对位有绿色色块,以大体积基团取代可以增加化合物的活性;邻位有黄色色块,无基团或小基团取代可以得到更好的活性;Cα部位二氯苯环取代基上的大面积绿色色块预示着在此增加立体位阻同样能够提高化合物的活性。图5(c)是CoMSIA模型静电场的三维等势图,红色色块表示在此处应引入负电性基团,而蓝色色块则表示此处增加正电性基团对活性的提高更有利。从图中可以看出,异羟肟酸部分的取代苯环上有大面积的红色色块,说明使用负电性基团对此苯环进行取代能有效地提高化合物活性,如-F和-CF3等。Cα部位的二氯苯环取代基下方存在大片红色色块,尝试用电负性更大的基团取代氯原子或许可提高活性。图5(d)是CoMSIA模型氢键受体场的三维等势图,洋红色色块表示在此处增加氢键受体可提高化合物活性,而红色色块则表示应在此处减少氢键受体的存在。从此图上可以看到,Cα部位的二氯苯环取代基下存在大面积的洋红色色块,说明此处引入氢键受体能有效的提高化合物的活性,如含氧杂环取代的化合物10(pIC50=5.824)的活性大于萘取代的化合物11(pIC50=5.237),有吡啶环取代的化合物17(pIC50=5.481)和15(pIC50=6.097)的活性大于仅用苯环取代的化合物12(pIC50=5.387),因此后续的结构改造也应遵循此方向。
2.3 分子对接结果及分析
分子对接结果和结合模式分析以拥有最高抑制活性的27号化合物为模板进行展示,可以看到,整个分子被蛋白活性口袋紧紧包裹,如图6所示。异羟肟酸部分的羟基氧朝向Mn2+,方便与金属离子形成紧密的配位作用,此特征相互作用在以往的研究里也有报道[20],说明本研究使用的分子对接协议可靠性较高。另外,异羟肟酸部分的取代苯环探向结合口袋深处,对其间位和对位进行大体积基团取代能使小分子与蛋白活性部位结合得更加牢固,此结论与3D-QSAR的结果相吻合,进一步验证了所建模型的可靠性。
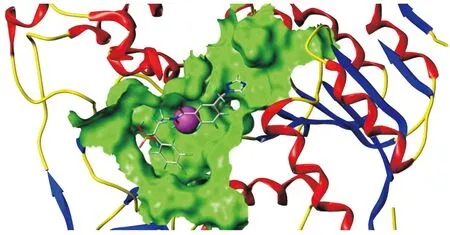
绿色区域为蛋白对接口袋,灰色棍为27号化合物分子,紫色圆球为Mn2+
mtDXR蛋白活性部位与27号化合物分子的非键作用分别用三维图(图7)和二维图(图8)展示,以便更好地分析其相互作用。

红色细棍为关键氨基酸残基,灰色粗棍为27号化合物分子,绿色虚线表示氢键,灰色虚线表示金属配位作用,淡紫色虚线表示疏水作用,淡绿色虚线表示非典型氢键作用

图8 27号化合物与mtDXR蛋白相互作用二维展示图
首先,27号化合物分子磷酸基上的羟基氧与氨基酸残基SER177形成了1个强氢键,羰基氧与氨基酸残基SER245形成了一个强氢键,距离分别为1.98 Å和1.89 Å。异羟肟酸部分的羟基氧与氨基酸残基GLU153形成了一个距离为2.88 Å的强氢键,而三氮唑上的氮原子也与氨基酸残基LYS128形成了一个距离为2.12 Å的强氢键。氢键的生成大大增加了化合物分子与受体结合的牢固性。其次,异羟肟酸部分的羟基氧与二价金属离子锰形成了O-Mn键(1.88 Å),此金属螯合作用有助于配体分子与蛋白靶点的紧密结合。再次,疏水作用广泛存在,氨基酸残基MET267与异羟肟酸部分的取代苯环以及二氯苯环上的氯原子皆形成了较强的疏水相互作用,距离分别为5.47 Å、4.14 Å和5.18 Å。二氯苯环上的对位氯原子与氨基酸残基PRO265形成了距离为4.86 Å的弱疏水相互作用,若尝试在此处用一个疏水性较强的基团取代,可能会增强与氨基酸残基的疏水作用进而增加整体化合物的活性,如-Br。三氮唑取代基也与氨基酸残基LYS128以及ALA126形成了弱的疏水相互作用,距离分别为5.38 Å和4.74 Å。另外,二氯苯环上的对位氯原子附近有氨基酸残基HIS248,并且形成了一个弱氢键(2.80 Å),在此处增加氢键受体能加强氢键相互作用,从而提高整体化合物活性,而且由于组氨酸在生理条件下是正电荷氨基酸,因此,在苯环上用负电性基团取代氯原子有利于化合物活性的增加,此结论与CoMSIA模型的氢键受体场和静电场的分析结果相吻合。
2.4 全新膦胺霉素衍生物的设计及评价
结合3D-QSAR的三维等势图和分子对接结果,可以确定膦胺霉素类小分子化合物的结构修饰区域,然后以原抑制活性最高的27号化合物分子为模板,进行结构的优化和改造,以得到拥有更高预测活性的新化合物分子。通过对静电场、立体场和氢键受体场的分析,以及分子对接显示的配体与受体活性部位关键氨基酸残基的相互作用模式,在保留膦胺霉素基本骨架的同时,异羟肟酸部分的取代苯环的间位和对位上应该引入大体积且负电性的基团,如-F、-CF3;二氯苯环处可引入疏水性更高的基团取代氯原子,如-Br;二氯苯环处还可引入电负性较大的氢键受体取代氯原子,如-COR、-OAr等,同时也满足了此处需要大体积基团占位的需求。
根据此思路,本文设计出了14个全新的膦胺霉素类小分子化合物(图9),其结构和预测活性如表4所示。利用已建立的CoMFA和CoMSIA模型分别预测这14个新化合物的活性,并且根据已有的分子对接协议对其进行打分,然后和原最高抑制活性的27号化合物相比较,结果显示:新化合物的对接得分和预测活性普遍较高,特别是化合物27m,同时拥有比27号化合物更高的对接得分和预测活性,说明上述的改造策略取得了初步成功,通过增强膦胺霉素类小分子化合物和mtDXR蛋白的非键作用,提高了配体受体的结合牢固性及化合物的整体抑制活性。化合物27m也可作为膦胺霉素类mtDXR抑制剂的先导化合物,进行进一步的评价和研究。

图9 膦胺霉素衍生物的分子结构示意图
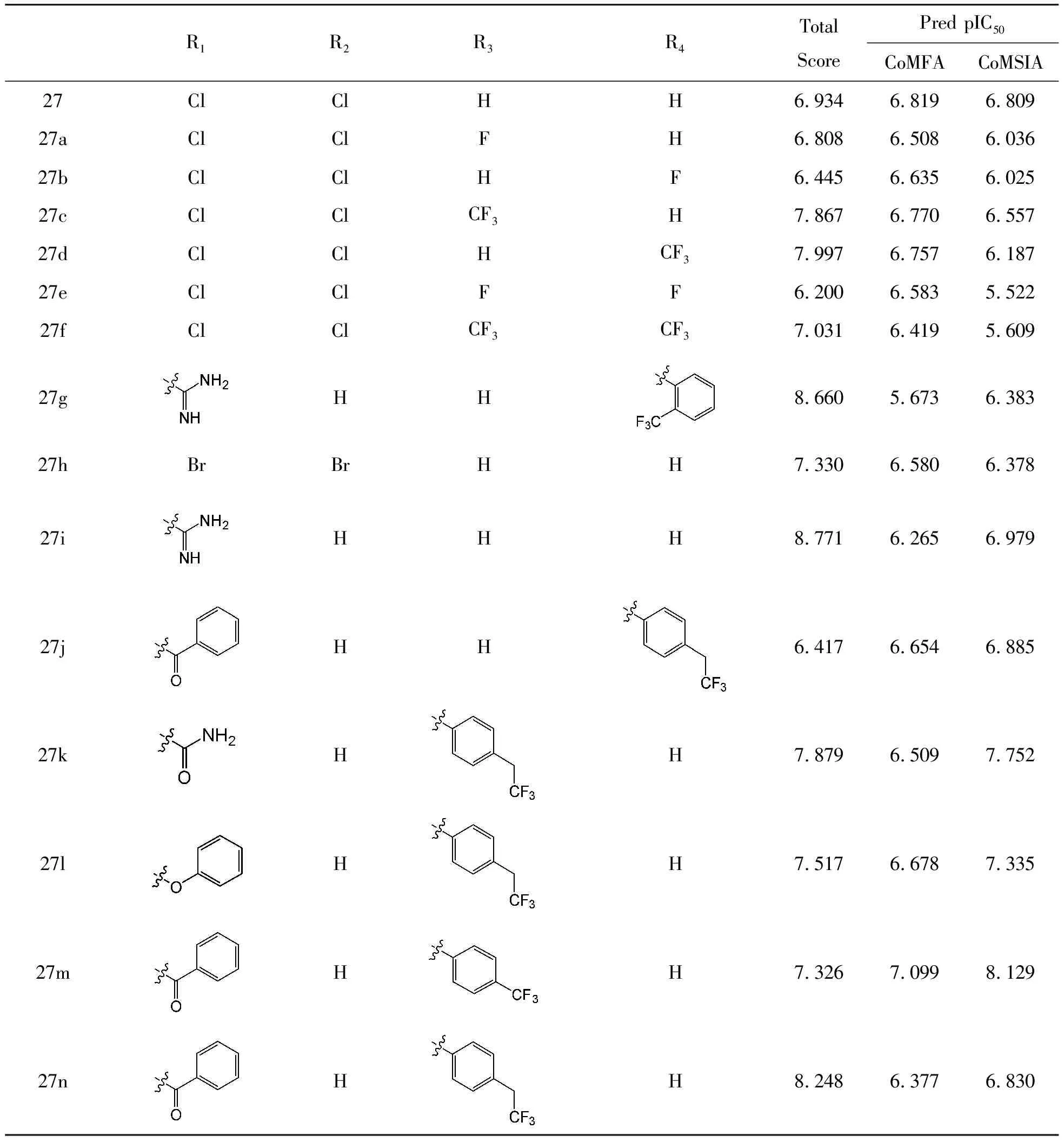
表4 新设计膦胺霉素衍生物的结构和预测活性
3 结论
结核病作为一种威胁着人类健康的全球性传染病,随着细菌耐药性的增加亟需新的治疗策略。本研究以结核分枝杆菌1-脱氧-D-木酮糖-5-磷酸还原异构酶(mtDXR)为抗菌靶点,收集了35个对其有抑制活性的膦胺霉素类小分子化合物,利用它们建立了3D-QSAR模型,分析了小分子抑制剂的结构和其生物活性之间的定量构效关系,结合CoMFA和CoMSIA模型的立体场、静电场和氢键受体场等势图,确定了膦胺霉素类小分子骨架上两大部位基团的改造策略,即异羟肟酸部分取代苯环的间位和对位上引入大体积且负电性的基团,以及Cα部位的二氯苯环取代基处引入电负性较大的氢键受体。同时,利用已知的靶点蛋白结构以及分子对接技术,揭示了小分子抑制剂和靶点蛋白活性位点的结合模式,并分析了小分子抑制剂和关键氨基酸残基之间的氢键作用、疏水作用以及金属螯合作用等,从分子层面验证了3D-QSAR的基团改造策略,并且提供了更深层次的指导和参考。最后,依据上述思路设计出了14个新型膦胺霉素类小分子化合物,对它们进行了分子对接打分和活性预测,得到了拥有更高对接分数和预测活性的膦胺霉素类小分子化合物27 m,可供作为先导化合物进行进一步的研究,而本文的研究也为后续的mtDXR小分子抑制剂的合理设计提供了可靠的理论基础。
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
发明与创新·小学生(2023年8期)2023-06-22 09:53:50
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
通信技术(2019年12期)2019-12-11 02:23:54
陶瓷科学与艺术(2016年1期)2016-02-28 16:33:11
池州学院学报(2015年3期)2016-01-05 01:13:04
小雪花·成长指南(2015年10期)2015-10-23 08:42:15
天津科技大学学报(2015年2期)2015-08-09 01:40:42
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
