
误诊为重症肌无力的MT-ATP6基因突变相关晚发性Leigh综合征2例及文献复习*
2023-03-10 01:58杨作臻
重庆医学 2023年4期
袁 梦,陈 俊,张 佳,杨作臻,甘 靖△
(1.四川大学华西第二医院儿科/出生缺陷与相关妇儿疾病教育部重点实验室,成都 610041;2.发育与妇儿疾病四川省重点实验室,成都 610041;3.宜宾市第二人民医院儿科,四川宜宾 644000;4.赛福解码(北京)基因科技有限公司,北京 100089)
Leigh综合征(LS)是一种由线粒体氧化磷酸化缺陷造成的进行性神经退行性疾病[1-2]。常见临床表现包括喂养困难、肌张力障碍、共济失调、眼肌麻痹、构音障碍、癫痫发作、精神运动发育迟缓等,伴血液、脑脊液、尿液中乳酸(Lac)增高[3]。LS预后差,患儿常于症状出现后2年内死亡,晚发性LS较为罕见,病程相对缓和,但由于其临床表现缺乏特异性,常易被误诊为其他导致肌无力的疾病[4]。本研究对2例被误诊为重症肌无力的晚发性LS患儿临床资料进行回顾性分析并复习相关文献,旨在为该类患儿的诊疗提供经验。
1 临床资料
1.1 基本病史
例1,男,13岁4个月,因“双侧眼睑下垂1个月”入院。眼睑下垂伴眼睑水肿、复视、眼痛,晨轻暮重,活动耐量稍下降,外院诊断“重症肌无力(眼肌型)”,给予溴吡斯的明及激素治疗但效果欠佳。既往史及个人史无特殊情况。家族史:患儿弟弟及表妹均于9岁左右患“呼吸道感染”后猝死;患儿表弟4岁时被开水烫伤,随后出现高热、语言和运动明显倒退(此前生长发育正常),不能行走、说话。基因检查提示MT-ATP6基因突变(m.9176T>C),考虑LS,给予“鸡尾酒疗法”治疗后语言、运动功能逐渐好转,目前8岁龄,运动正常,语言稍落后;患儿父母体健。查体:步态正常,视力、视野粗测正常,双侧眼睑下垂,瞳孔等大等圆,对光反射灵敏,眼球活动正常,四肢肌力、肌张力正常。辅助检查:抗乙酰胆碱受体免疫球蛋白G(IgG)抗体阳性(0.95 mmol/L);新斯的明试验、疲劳试验、重复电刺激、脑电图结果均正常;血Lac、脑脊液Lac检测值均在正常范围内。头颅磁共振成像(MRI)示:导水管周围脑白质区纵行条片状异常信号,双侧背侧丘脑和基底节区片状异常信号。磁共振波谱成像(MRS)示:导水管周围脑白质呈N-乙酰天门冬氨酸(NAA)/肌酸(Cr)减低,可见明显的Lac峰。见图1。
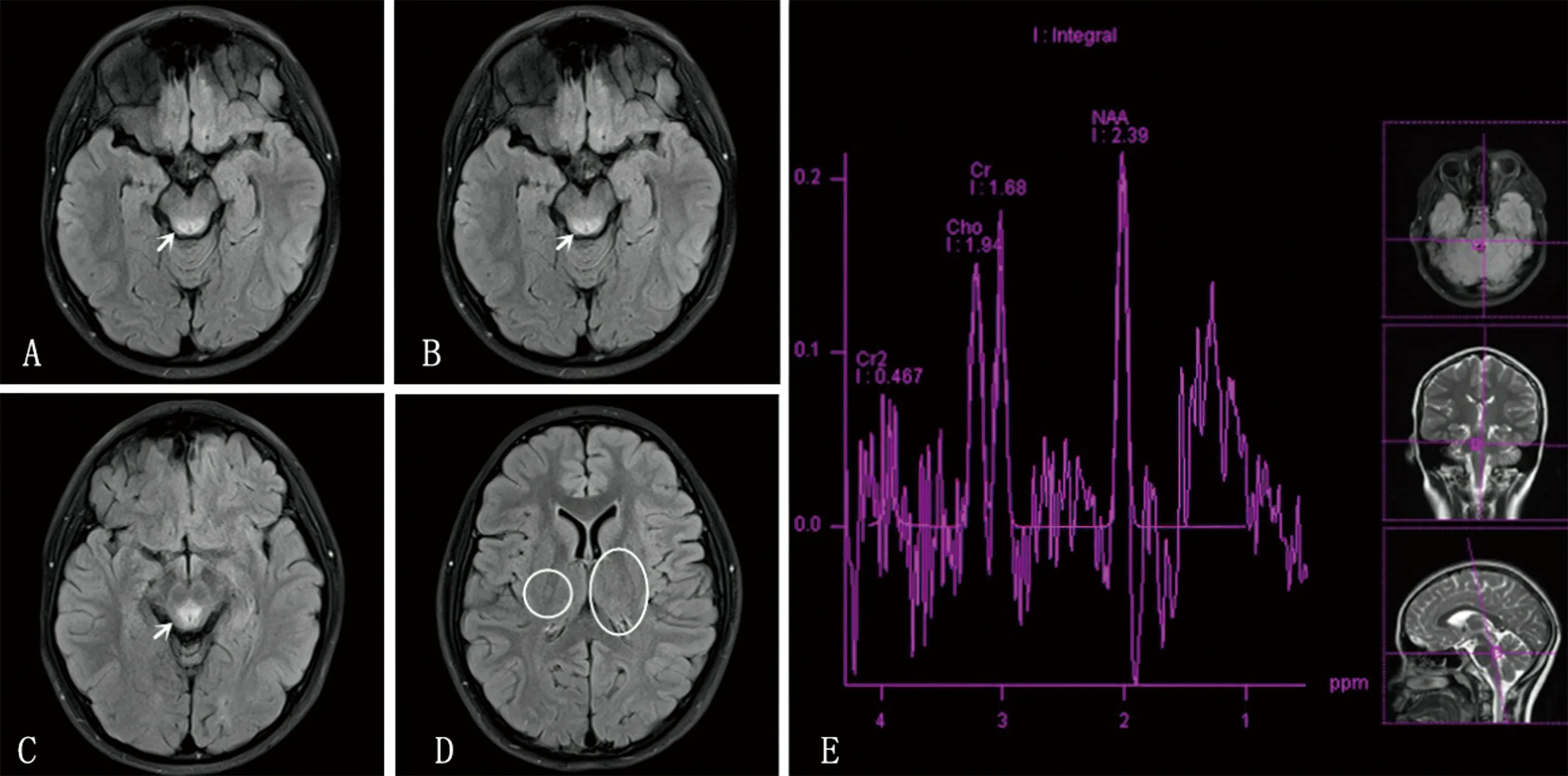
A、B、C、D:头颅MRI图像,白色箭头示导水管周围脑白质区纵行条片状异常信号,白色圆框示双侧背侧丘脑和基底节区片状异常信号;E:MRS图像。
例2,女,9岁11个月,因“双侧眼睑下垂、复视6 d”入院。眼睑下垂,晨轻暮重,伴复视、眼球活动受限。门诊完善相关检查考虑“重症肌无力(眼肌型)”,予溴吡斯的明治疗但效果欠佳。既往史、个人史、家族史无特殊情况。查体:步态正常,双侧眼睑下垂,双侧眼球向上运动活动稍受限,复视、瞳孔等大等圆,对光反射灵敏,四肢肌力、肌张力正常。辅助检查:新斯的明试验呈可疑阳性;抗乙酰胆碱受体IgG抗体阳性(0.92 mmol/L);疲劳试验、重复电刺激、脑电图结果均正常。入院后查血Lac升高(2.92 mmol/L)、脑脊液Lac升高(3.64 mmol/L)。头颅MRI示:中脑导水管周围组织、桥脑背侧和延髓有斑片样信号异常,以中脑导水管周围组织信号异常显著。MRS示:倒置的Lac峰稍明显,提示遗传代谢性疾病可能性大。见图2。
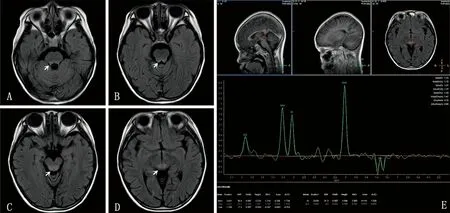
A、B、C、D:头颅MRI图像,白色箭头示中脑导水管周围组织、桥脑背侧和延髓有斑片样信号异常;E:MRS图像。
1.2 基因检测方法
1.2.1高通量靶向测序
抽取先证者及父母外周血2 mL,使用试剂盒(北京天根生化科技有限公司)提取外周血DNA,采用Qubit定量后,使用超声破碎仪将DNA打碎至150~350 bp,进行聚合酶链式反应(PCR)扩增和文库构建。采用覆盖全外显子和线粒体基因的panel(iWES,北京赛福解码基因科技有限公司)进行捕获建库,采用双末端(Paired-End)测序策略。Raw data>10 G,Q30≥80%,确保线粒体基因区域测序深度达到8 000×以上。
1.2.2生物信息学分析和Sanger测序验证
测序仪下机原始数据使用BWA软件比对人类参考基因组GRCh38/hg38,采用GATK进行变异检出。使用Annovar工具对.vcf变异文件进行注释。致病变异位点筛选:(1)筛选出外显子区和线粒体区域变异、非同义变异位点;(2)HGMD、gnomAD等数据库中未见或携带率小于5%;(3)参考dbSNP、OMIM、HGMD、ClinVar等多种数据库对致病变异位点进行评估;(4)使用SIFT、Polyphen2、LRT、MutationTaster等多种预测软件进行基因突变打分预测。
1.2.3变异的致病性分析
根据美国医学遗传学与基因组学学会(ACMG)遗传变异分类标准与指南,对变异的致病性进行判定。
1.3 基因检测结果
2例患儿均检出MT-ATP6基因存在m.9176T>C突变(图3),均为均质突变(即突变后绝大部分线粒体基因的序列一致),突变比例分别为99.4%和100.0%。由于例1患儿家系中的表弟有明确的位点突变,作者对其家系进行线粒体基因突变验证(图4),发现该患儿突变来源于外婆,突变比例为21.19%;母亲突变比例为97.95%,父亲为野生型;小姨突变比例为95.50%,表弟(小姨的儿子)突变比例为99.89%,舅舅突变比例为57.44%。例2患儿变异来源于母亲,其母亲表型正常。

A:例1患儿MT-ATP6基因突变(m.9176T>C)图,变异来源于其母亲;B:例2患儿MT-ATP6基因突变(m.9176T>C)图,变异来源于其母亲。

图4 例1患儿家系图
1.4 诊疗经过及预后
根据临床表现及实验室检查、基因检查结果,明确LS诊断后,参考国内外报道的MT-ATP6基因突变(m.9176T>C)所致晚发性LS的诊疗情况(表1),对2例患儿采用“鸡尾酒疗法”(维生素B1,10 mg·kg-1·d-1;维生素B2,10 mg·kg-1·d-1;维生素C,10 mg·kg-1·d-1;维生素E,10 mg·kg-1·d-1;左卡尼汀,50 mg·kg-1·d-1;辅酶Q10,10 mg·kg-1·d-1)改善代谢。例1患儿治疗10 d后双侧眼睑下垂恢复,其他临床表现正常;例2患儿治疗3个月后,眼睑下垂及复视明显改善,但在长时间活动后仍有肢体乏力表现,复查血Lac为1.29 mol/L,较此前明显下降。

表1 MT-ATP6基因突变(m.9176T>C)相关晚发性LS患者临床资料
2 讨 论
LS具有显著的临床表型和遗传异质性,其遗传方式包括常染色体隐性遗传、X连锁遗传、母系遗传,其中核基因突变占70%~80%,线粒体基因(mtDNA)突变占10%~20%[12]。ATP合成酶6(ATPase6)缺陷可导致LS,其编码基因为MT-ATP6基因(OMIM 516060),位于mtDNA 8527-9207,目前已发现多种MT-ATP6基因突变所导致的LS,包括:m.8993T>C、m.8993T>G、m.9176T>C、m.9176T>G、m.9185T>C、m.8597T>C、m.9035T>C、m.8936T>A、m.9191T>C[11,13-19]。mtDNA与核DNA在突变遗传和致病种类上有较多区别,其遗传特性包括母系遗传、高突变发生率、均质突变与异质突变相互变化、能量阈值效应和瓶颈效应、随细胞衰老突变发生率增加等。mtDNA突变的异质性普遍存在,目前已明确阈值效应对mtDNA异质突变致病性的影响,当突变发生率达到一定的阈值后,患者可出现相应的临床表型。临床普遍认为,mtDNA突变发生率>70%可达致相应的临床表型[20]。在MT-ATP6异质突变中,突变发生率>90%可导致LS和代谢性中风;在较低的突变水平时,患者可出现神经病变、共济失调和视网膜色素变性[21],但目前尚无其具体阈值的进一步研究报道。在作者所报道的例1患者家系中,先证者和其表弟、母亲、小姨的突变比例均>95%,其中先证者和表弟(先证者小姨的儿子)突变发生率达99%以上,均出现了临床表现,而突变发生率稍低的先证者母亲、小姨目前表型均正常,可能与m.9176T>C位点突变相关LS发病较晚、症状相对缓和有关。另外,先证者弟弟及表妹(先证者小姨的女儿)均患“呼吸道感染”后猝死,虽未行基因检测,但根据家系患病情况推测,两者均有极大可能也携带有MT-ATP6基因突变(m.9176T>C)。因此,作者猜测对于该位点的阈值效应突变发生率可能>99%。
在MT-ATP6基因突变所致的LS中,以早发性更为多见,发育迟缓或倒退、共济失调是常见的起始临床表现[22],但m.9176T>C位点突变导致的LS则以晚发多见。作者所报道的2例病例突变位点均为m.9176T>C,该位点突变已报道共15例[5-11,23-25],其中7例为早发性LS,于2岁前起病(9个月至1岁9个月);8例为晚发性LS,于2岁后起病(2~27岁)。本研究2例患儿分别于13岁4个月、9岁11个月起病,以眼睑下垂为主要临床表现,这与OH等[5]报道的病例表现一致。不同的是,作者报道的2例患儿在新斯的明试验、抗乙酰胆碱受体IgG抗体检查中均提示阳性,而被误诊为重症肌无力(眼肌型),治疗效果均不佳。由此可见,晚发性LS的临床表现可能不典型,容易误诊为重症肌无力或其他以肌无力为首发症状的疾病,临床可根据头颅MRI和基因检测进行区分。
目前,LS大多采用对症治疗,重点是改善能量状态和降低Lac水平,包括补充辅酶Q10、多种维生素(鸡尾酒疗法)、丙酮酸、二氯乙酸,以及生酮饮食等[12]。对于m.9176T>C位点突变导致的晚发性LS患者,也有使用血浆置换及免疫球蛋白治疗有效的病例,但其机制尚未明确[9]。雷帕霉素、低氧疗法、腺病毒相关病毒介导的基因治疗也有望在将来成为LS的有效治疗方法。“鸡尾酒疗法”是LS在临床中最常采用的治疗方法,对表1中8例(包括本研究2例患者及例1患者表弟)采用“鸡尾酒疗法”治疗的患者进行分析,所有患者均为晚发性LS,其中1例患者死亡,可能与其启动“鸡尾酒疗法”时间较晚(起病2个月后)有关,另外7例患者存活至今,表明“鸡尾酒疗法”治疗m.9176T>C位点突变的晚发性LS患者有效。WEI等[11]报道了16例线粒体基因突变所致的晚发性LS患者,其中11例患者接受了“鸡尾酒疗法”并随访超过1年,结果显示5例患者病情稳定,病变减少。因此,早期明确诊断并采用“鸡尾酒疗法”,能够有效改善晚发性LS患者临床症状及预后。
综上所述,MT-ATP6基因突变(m.9176T>C)导致的晚发性LS更为多见。晚发性LS临床表现不典型,常以眼肌无力起病,早期易误诊为重症肌无力,头颅MRI和基因检测是诊断该类患者最有效的手段。“鸡尾酒疗法”能够改善晚发性LS患者的临床症状和预后,阻止或减缓疾病进一步进展。
猜你喜欢
中老年保健(2021年12期)2021-08-24
皮肤病与性病(2021年3期)2021-07-30
中学生数理化·八年级物理人教版(2020年12期)2021-01-18
世界科学技术-中医药现代化(2020年2期)2020-07-25
当代水产(2020年4期)2020-06-16
国际放射医学核医学杂志(2020年2期)2020-05-30
现代装饰(2020年3期)2020-04-13
现代装饰(2019年11期)2019-12-20
中国中医急症(2019年10期)2019-05-21
基层中医药(2018年3期)2018-05-31
