
氧空位和磷掺杂协同增强铁钴基材料电解水催化性能
2023-02-27 03:29曾传旺李潇潇曾金明赖嘉俊漆小鹏
无机化学学报 2023年2期
曾传旺 李潇潇 曾金明 刘 超 赖嘉俊 漆小鹏*,
(1江西理工大学稀土学院,赣州 341400)
(2赣州市氢能材料与器件重点实验室,赣州 341000)
0 引言
目前,全球处于常规能源(石油、煤炭和天然气等)与清洁能源(太阳能、生物质能和氢能等)迭代的阶段[1]。随着人口和经济的持续快速增长,寻找无污染、可持续、能够在全球范围内通用的替代能源已经成为一项紧迫和具有挑战性的任务[2]。氢因其独特的可再生性、丰富性、高能量密度、生产来源多样、无污染等特性,被认为是最富潜力的新一代能源载体[3]。通过电解水制得的氢,可以作为未来绿色可持续发展的主要氢源之一[4]。电解水过程具有较高的热力学势垒和缓慢的动力学特性,总是需要高效的电催化剂来降低输入电位[5]。在工业中使用的最常见的催化剂大多是基于贵金属(Pt、Ir、Ru),然而它们的高价格和稀缺性阻碍了其在电解水中的广泛应用[6]。因此,寻找储量丰富、低成本、高活性的非贵金属电催化材料是目前电解水制氢的热点[7]。
过渡金属复合物(TMCs)可靠性高、成本低,受到众多研究者的青睐。其中,钴基材料因其来源丰富、种类多,在过渡金属化合物(钴、铁、镍、钨、锰等)中尤为重要。钴基材料具有不同的价态(Co2+/Co3+),金属氧化还原电位接近1.23 V(vs RHE),被视为高效的析氧反应(OER)催化剂[8]。氧空位(Ov)的引入可以有效调控催化剂表面的电子结构,优化反应物在催化剂表面的吸附能,从而降低反应能垒,有利于O2分子的吸附。例如,Zhang等[9]采用水热法制备钴基前驱体,然后预氧化制备 Ovac‑Co3O4‑T‑t。经过探索发现,Ovac‑Co3O4‑320‑5由于超薄的纳米片结构和氧空位的存在,既增强了活性,又提高了转移速率,展现出较好的OER活性。研究表明,与单一的氧化物(Co3O4、MnO2、Fe3O4等)比 较 ,双 金 属 基 氧 化 物(CoFe2O4、CuFe2O4和 NiFe2O4等)氧化还原性能优越,具有较高的催化活性,是OER的重要候选物[10‑11]。CoFe2O4已被报道为一种很有前途的OER和氧还原反应(ORR)电催化剂[12],然而CoFe2O4在析氢反应(HER)中表现出电导率差、动力学过程缓慢的特性,需要较高的析氢过电位,因此不利于作为双功能或多功能电催化剂[13]。非金属掺杂(S、P、N和Se等)材料现已被证明是性能优异的水裂解电催化剂[14]。在这些材料中,过渡金属磷化物表现出卓越的析氢性能,结合前期研究及查阅文献发现:(1)在水裂解过程中,金属磷化物(M‑P)中的磷可以通过与反应中间体的适度键合来参与反应,在其表面构成质子/氢化物受体位点,制备高活性材料[15];(2)与单金属的磷化物比较,双金属的磷化物由于不同材料的协同调控效应而展现出更卓越的析氢性能[16];(3)引入多种金属,不仅提高了不同金属离子之间的电荷转移速率,还改变了双金属材料的电子结构,从而能大大降低其在电催化过程中的动力学势垒[17]。综上所述,氧空位增强了催化活性并使电子转移速度更快,而磷掺杂可以调整其表面电荷状态,这最终促进了HER和OER性能。本研究提出将氧空位和磷掺杂结合起来,协同增强铁钴基双金属材料电催化性能,构建更高效的电解水催化体系。
1 实验部分
1.1 试 剂
无水乙醇(C2H5OH)、六水合硝酸钴(Co(NO3)2·6H2O)、次亚磷酸钠(NaH2PO2)购自西陇科学股份有限公司;浓盐酸(HCl)、氢氧化钾(KOH)购自国药化学试剂有限公司;以上试剂均为分析纯。泡沫铁(IF)购自探乾郎新材料公司。
1.2 实验过程
1.2.1 铁钴基前驱体的制备
第一步,将商用的泡沫铁裁剪成1 cm×2 cm的长方形,无水乙醇超声20 min,去除表面杂质,然后用水清洗。第二步,将处理过的泡沫铁浸泡在3 mol·L−1HCl中,15 min后取出,用水冲洗。同时,配制硝酸钴溶液,称取x克(x=0.145 7、0.291 3、0.436 9)硝酸钴,加入8 mL超纯水,搅拌40 min,得到粉红色溶液。第三步,将3片处理好的泡沫铁放入盛有上述溶液的烧杯中,将烧杯放置在25℃房间内,静置24 h。最后,冲洗负载静置产物的表面,将所得产物放置真空干燥箱,静置一夜,获得前驱体。
1.2.2 样品Ov‑CXFO/IF(X=L、M、R)的制备
将含有不同质量钴(X=L、M、R,分别对应0.145 7、0.291 3、0.436 9 g)的前驱体垂直置于陶瓷舟皿中,然后将舟皿放置于石英管(壁厚3 mm)的中心。首先将石英管抽至真空状态,随后通氩气至正常气压,然后再将石英管抽至真空状态,真空泵一直运作,保证石英管内为负压、无氧。温度编程:在80 min升至400℃,不保温,自然冷却,然后取出,即可获得Ov‑CXFO/IF样品。
1.2.3 样品CMFO/IF的制备
取x=0.291 3的前驱体垂直置于陶瓷舟皿中,然后将舟皿放置于石英管(壁厚3 mm)的中心。在空气的气氛下,炉子从20℃升至400℃,用时80 min,不保温,自然冷却,即可获得CMFO/IF样品。
1.2.4 样品P‑Ov‑CXFO/IF的制备
称取1.2 g NaH2PO2放入特定的陶瓷舟皿内,然后将分别装有Ov‑CXFO/IF和NaH2PO2的陶瓷舟皿并排放入专门磷化的石英管中,NaH2PO2的舟皿置上游,Ov‑CXFO/IF的舟皿放下游,间隔大约2.5 cm。在氩气的气氛下,炉子从20℃升至400℃,用时80 min,保温1 h,自然冷却,即获得P‑Ov‑CXFO/IF样品,制备过程如图1所示。

图1 P‑Ov‑CMFO/IF的制备流程Fig.1 Synthesis process of P‑Ov‑CMFO/IF
1.2.5 样品P‑Precursor/IF的制备
P‑Precursor/IF的磷化方法和P‑Ov‑CMFO/IF 类似,但直接磷化前驱体,没有真空热处理这一步骤。
1.2.6 Pt/C和RuO2材料的制备
称取2.035 mg的20%Pt/C放入1 mL的试管中,然后依次加入乙醇(360 µL)、超纯水(120 µL)、Nafion(20µL)。将混合液放置超声机超声55 min,最终得到均匀的黑色液体,然后将黑色液体分次逐滴滴入泡沫铁表面,等待其干燥。RuO2的合成与20%Pt/C制备相似,称取同等质量的RuO2取代20%Pt/C。
1.3 表征手段及电化学测试
XRD原始数据由Bruker D/max‑2500(日本理学)高级X射线衍射仪(Cu Kα,λ=0.154 nm;2θ范围10°~90°;管电压40 kV;管电流40 mA)收集。用∑IGMA扫描电子显微镜(SEM,德国卡尔蔡司公司)观察所制备的纳米花形貌,工作电压为15kV。透射电子显微镜(TEM)、能量色散X射线谱(EDS)和高分辨率TEM(HR‑TEM)都是在电压为200 kV的Talos F200x(美国辉门公司)上进行的。采用X射线光电子能谱(XPS,Thermo Scientific K‑Alpha,赛默飞世尔科技公司;I=15 mA,U=15 kV,hν=1 486.6 eV)研究所制备的样品化学特性和氧空位。
所有制备好的催化剂的电化学测试均使用三电极系统完成,并使用电化学工作站(CHI760E,上海辰华仪器有限公司)测试。在KOH电解液(1.0 mol·L−1,pH=14)中,以制备的催化剂(1 cm×1 cm)为工作电极,碳棒为对电极,Ag/AgCl(KCl溶液)为参比电极。使用线性扫描伏安法(LSV)表征HER、OER和电解水反应,测量范围在−0.8~−1.6 V、0~1 V和0~2 V(vs Ag/AgCl),速率为 2 mV·s−1。电化学阻抗谱(EIS)在110 mV的过电位下进行了理想测量,频率范围:0.01 Hz~100 kHz。
2 结果与讨论
2.1 材料的微观形貌
图 2A、2B为 P‑Ov‑CMFO/IF的 SEM 图片。从SEM图中可以看出泡沫铁上原位生长的铁钴基纳米花分布均匀,比较密集,经测量纳米花片层厚度约为42 nm,纳米花表面有许多小颗粒,这可能是由于加热造成的体积效应,使暴露面积更大[18‑19]。图2C为P‑Ov‑CMFO/IF的TEM图,可以看出纳米花的片层比较薄,纳米孔大小为2~5 nm(红色圆圈),这可能是PH3气体腐蚀的效果。此外,如图2D所示,P‑Ov‑CMFO/IF还存在CoFe2O4和CoP晶格条纹,间距为0.210和0.242 nm,对应CoFe2O4的(400)面和(222)面;间距为0.230 nm,对应CoP的(201)面。高角环形暗场‑STEM(HAADF‑STEM)图、EDS谱图(图 2E)和元素映射图(图2F)显示,Fe、Co、O和P在P‑Ov‑CMFO/IF均匀分布,从EDS谱图中可以看出P‑Ov‑CMFO/IF中Co、Fe、O、P的原子百分比分别为26.57%、40.86%、15.32%、17.25%。由上可知,P成功掺入Ov‑CMFO/IF,并以CoP形式存在。

图2 P‑Ov‑CMFO/IF的SEM图像 (A、B)、TEM图像 (C、D)、HAADF‑STEM图和EDS谱图 (E)、元素映射图 (F)Fig.2 SEM images(A,B),TEM images(C,D),HAADF‑STEM image and EDS spectrum(E),and element mappings(F)of P‑Ov‑CMFO/IF
2.2 材料的结构分析和化学组成
为了进一步确定催化剂的组成,利用XRD对样品进行测试,图3为制备的Ov‑CMFO/IF、P‑Precursor/IF、P‑Ov‑CMFO/IF的XRD图。XRD图中2θ=44.68°、65.03°、82.34°处的衍射峰,归属于Fe的(110)、(200)和(211)三个晶面,为基底峰,峰强度较高。真空烧结(400 ℃)后,前驱体转化为 CoFe2O4,在 30.08°、35.44°、37.06°和 62.59°的衍射峰对应于 CoFe2O4的(220)、(311)、(222)和(440)晶面[15]。磷化后,引入磷源的 P‑Ov‑CMFO/IF 与 Ov‑CMFO/IF 相比,在 31.60°、48.13°、48.40°、56.78°和 77.99°的衍射峰,归属于CoP 的(011)、(211)、(202)、(301)和(204)晶面,但它们峰强较弱。由此可进一步说明,P成功掺入Ov‑CMFO/IF,并以CoP形式存在。
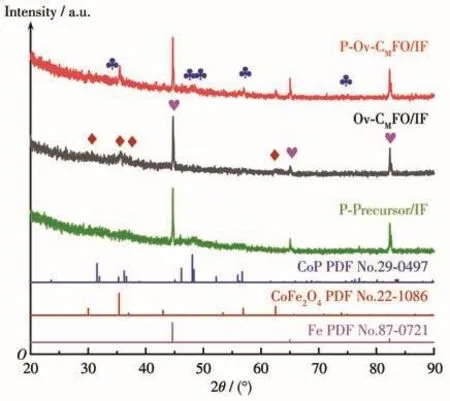
图3 P‑Ov‑CMFO/IF、Ov‑CMFO/IF和P‑Precursor/IF的XRD图Fig.3 XRD patterns of P‑Ov‑CMFO/IF,Ov‑CMFO/IF,and P‑Precursor/IF
为表征氧空位与P原子掺杂对于CoFe2O4的影响,用XPS对 Ov‑CMFO/IF和P‑Ov‑CMFO/IF进行分析。图4A显示了Ov‑CMFO/IF以及P‑Ov‑CMFO/IF的Co2p XPS谱图。对于P‑Ov‑CMFO/IF样品,结合能780.90 eV对应Co3+峰;结合能782.55 eV对应Co2+峰;结合能778.59和793.59 eV对应Co—P键。相较于Ov‑CMFO/IF样品,P‑Ov‑CMFO/IF的Co2+和Co3+峰位偏移0.85 eV,向高结合能移动。图4B显示了Ov‑CMFO/IF和P‑Ov‑CMFO/IF的Fe2p XPS谱图。对于P‑Ov‑CMFO/IF样品,结合能710.63和723.94 eV对应Fe2+峰;结合能713.07和726.18 eV对应Fe3+峰;结合能706.80 eV对应Fe—P键[20]。相较于Ov‑CMFO/IF样品,P‑Ov‑CMFO/IF的Fe2+和Fe3+峰位偏移0.46 eV,也向高结合能移动。在P‑Ov‑CMFO/IF的P2p XPS谱图中(图4D),包含2个典型双峰,结合能129.32和130.22 eV对应M—P键;结合能133.01和134.39 eV对应P—O键。综上所述,P掺入到了CoFe2O4中,改变了CoFe2O4的电子结构。
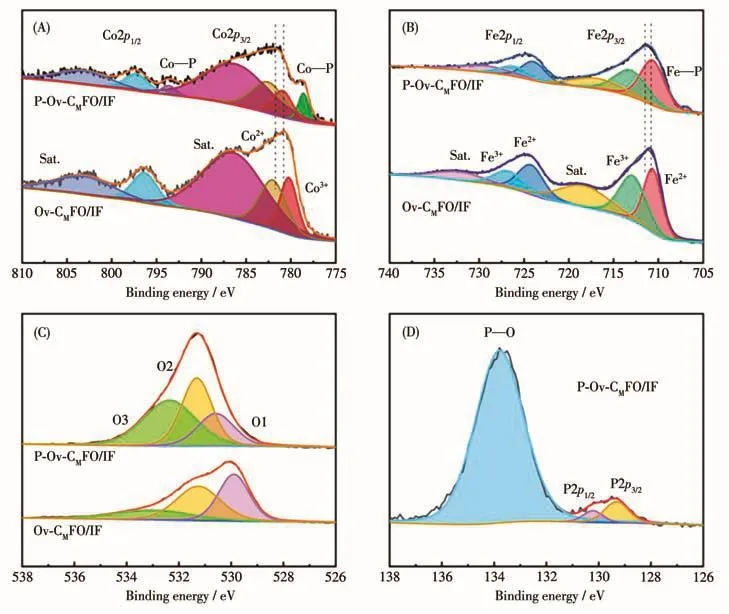
图4 Ov‑CMFO/IF和P‑Ov‑CMFO/IF的XPS谱图:(A)Co2p;(B)Fe2p;(C)O1s;(D)P2pFig.4 XPS spectra of Ov‑CMFO/IF and P‑Ov‑CMFO/IF:(A)Co2p;(B)Fe2p;(C)O1s;(D)P2p
图4C 显示了 Ov‑CMFO/IF和 P‑Ov‑CMFO/IF的O1s XPS谱图。对于 P‑Ov‑CMFO/IF样品,结合能532.36 eV对应于O3(羟基/吸附氧)峰;结合能531.32 eV对应于O2(氧空位)峰;结合能530.58 eV对应于O1(晶格氧)峰[21]。通过计算 Ov‑CMFO/IF 和 P‑Ov‑CMFO/IF中O2所占比例可得:Ov‑CMFO/IF中O2占41.18%,P‑Ov‑CMFO/IF中O2占38.91%。同时,由图4A计算Co2+与Co3+比例,对于Ov‑CMFO/IF和P‑Ov‑CMFO/IF,该比例分别为1.34和2.02,这说明在真空中Co3+还原为Co2+,诱导了氧空位的产生。
2.3 电化学析氢性能
为了评估氧空位、磷掺杂以及不同Co含量对材料电催化性能的影响,在 1.0 mol·L−1KOH(pH=14,25 ℃)中测试了7种催化剂(Ov‑CMFO/IF、P‑Ov‑CLFO/IF、P‑Precursor/IF、P‑Ov‑CMFO/IF、P‑Ov‑CRFO/IF、20%Pt/C和CMFO/IF)的电催化性能。7个样品的LSV曲线如图5A所示,在电流密度为−10 mA·cm−2时,P‑Ov‑CMFO/IF的过电位仅为54 mV。更为重要的是,在电流密度为−100 mA·cm−2时,P‑Ov‑CMFO/IF的过电位为119 mV,远远优于Ov‑CMFO/IF(345 mV)、P‑Precursor/IF(255 mV)和 CMFO/IF(352mV),说明氧空位和磷掺杂能协同增强材料电解水析氢性能。其余的几种材料 P‑Ov‑CLFO/IF、P‑Ov‑CRFO/IF 和20%Pt/C 在−100 mA·cm−2时过电位分别为 173、160、243 mV,说明钴的含量对其析氢性能也有影响。除此之外,在更大电流(400 mA·cm−2)下,P‑Ov‑CMFO/IF的过电势仅为191 mV,远远优于其余6种材料。综上所述,第一,含有磷和氧空位的样品(P‑Ov‑CMFO/IF)的性能优于仅含有氧空位(Ov‑CMFO/IF)和仅有P掺杂(P‑Precursor/IF)的样品,说明氧空位和磷掺杂协同增强了CoFe2O4电催化性能[23];第二,钴的含量对催化剂析氢性能也有影响,适量的钴才能展示出其优异的析氢性能。
根据LSV曲线获得Tafel斜率,了解合成样品的HER 机理。P‑Ov‑CMFO/IF 的 Tafel斜率(图 5B)为57.48 mV·dec−1,远远低于除 Pt/C(54.34 mV·dec−1)外的其余几种样品,说明制备的催化剂析氢反应遵循Volmer‑Heyrovsky机理[24]。如图5C所示,根据柱状图可更直观地看出P‑Ov‑CMFO/IF的HER性能优异,P‑Ov‑CMFO/IF有着最快的反应速率。
对上述除20%Pt/C和CMFO/IF以外的5种材料进行析氢阻抗EIS分析。由图5D计算得到,P‑Ov‑CMFO/IF 的 Rct最小(1.25 Ω),其他材料 Ov‑CMFO/IF、P‑Precursor/IF、P‑Ov‑CLFO/IF、P‑Ov‑CRFO/IF的Rct分别为 15.86、12.39、5.11、2.64 Ω。Rct越小,说明其电子转移速率越快,HER性能越好。综上所述,P‑Ov‑CMFO/IF的电子转移速率最快,HER性能最好。P‑Ov‑CMFO/IF的Rct小的原因:(1)P‑Ov‑CMFO/IF中的Ov减小带隙,加速转移;(2)磷的掺杂改变化合物周围电子,改善了其中的析氢动力学[20]。
另外,为探究制备材料的内在HER活性,根据除20%Pt/C以外的6种材料的循环伏安(CV)曲线(20~100 mV·s−1)计算其 Cdl。如图 5E 所示,P‑Ov‑CMFO/IF、Ov‑CMFO/IF、P‑Precursor/IF,P‑Ov‑CLFO/IF,P‑Ov‑CRFO/IF和CMFO/IF的Cdl分别为126.24、13.63、36.6、62.23、73和5.26 mF·cm−2。含有氧空位和P的P‑Ov‑CMFO/IF样品的Cdl,比真空后的Ov‑CMFO/IF和直接磷化的P‑Precursor/IF及不含氧空位的CMFO/IF样品高出数倍,表明P‑Ov‑CMFO/IF样品的电化学面积也比其余样品高出数倍[25]。综上所述,第一,P掺杂和氧空位既可以改善其电子结构,又增大其有效面积,使活性位点增多;第二,钴的含量对其有效面积也有影响,少量钴(P‑Ov‑CLFO/IF)和高量钴(P‑Ov‑CRFO/IF)材料的活性位点都较少。
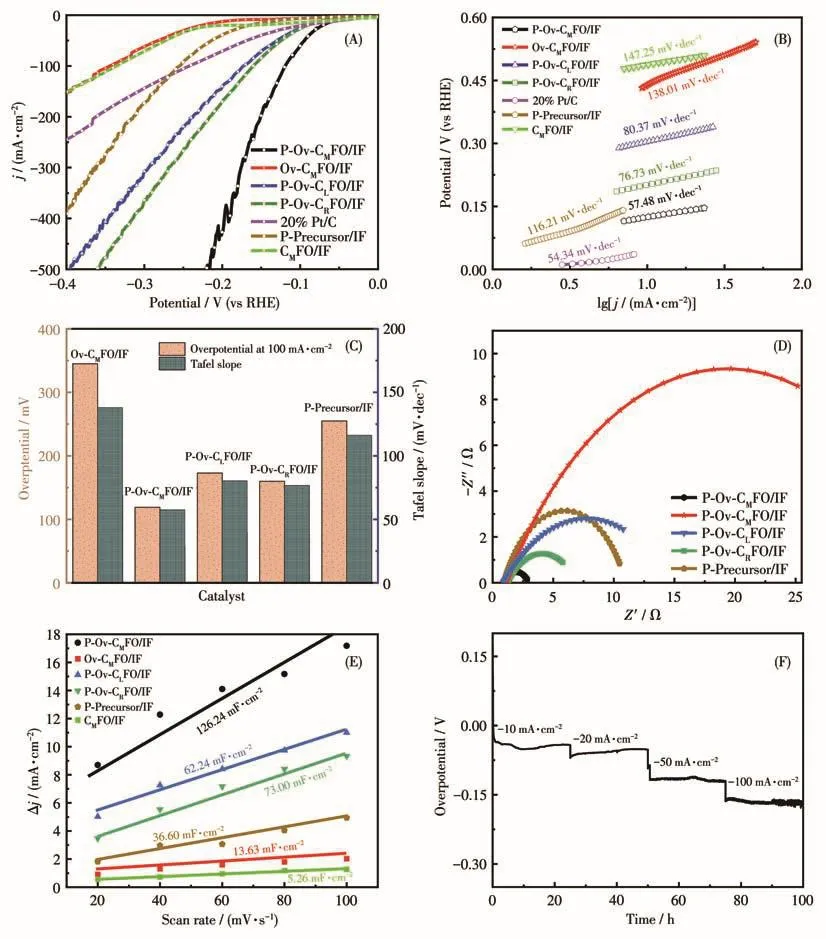
图5 不同样品的电催化性能:(A)LSV曲线;(B)Tafel图;(C)过电位和Tafel斜率柱状图;(D)EIS谱图;(E)Δj与扫描速率的关系;(F)P‑Ov‑CMFO/IF在不同电流下100 h的稳定性Fig.5 Electrocatalytic properties of different samples:(A)LSV curves;(B)Tafel diagram;(C)Histogram for overpotential and Tafel slop;(D)EIS spectra;(E)Relationship between Δj and scanning rate;(F)Stability of P‑Ov‑CMFO/IF at different current densities for 100 h
如图5F所示,连续100 h对P‑Ov‑CMFO/IF样品施加不同的电流(10、20、50、100 mA·cm−2),用来评价其HER稳定性。P‑Ov‑CMFO/IF在不同电流下其电压变化:在 10 mA·cm−2时,电压略有上升,然后下降,其中变化大约6 mV。在20 mA·cm−2时,电压略有下降,变化大约8 mV。在高电流50和100 mA·cm−2下的电压变化分别为10和11 mV。由上述可知,在不同电流下电压变化都可忽略不计,HER稳定性较好。
电化学比表面积(AECSA)是理想中电催化剂活性位点同电解液接触的面积。一般使用公式可以计算出AECSA。按照以下公式计算出AECSA,并用LSV曲线的电位值除以AECSA即可实现归一化处理[26]。
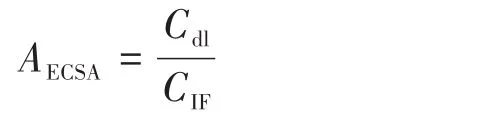
其中Cdl表示催化剂的双电层电容,其数值根据图5E计算;CIF表示的是泡沫铁的双电层电容,泡沫铁的双电层电容经测试为22.53 mF·cm−2。图6是对极化曲线ECSA归一化后的图像,从图中可以明显看出在相同电流密度下P‑Ov‑CMFO/IF对应的电压最小,表明氧空位和磷掺杂后的样品具有最高的本征活性。
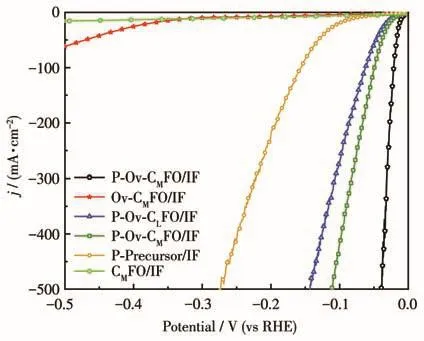
图6 不同样品的极化曲线归一化到ECSAFig.6 Polarization curves of different samples normalized to ECSA
2.4 材料的析氧和全解水电化学性能表征
为了研究 P‑Ov‑CMFO/IF 纳米花簇在 1.0 mol·L−1KOH中的OER性能,对除20%Pt/C外的7种材料进行性能测试。如图7A所示,在电流密度为10 mA·cm−2时,P‑Ov‑CMFO/IF 的析氧过电位仅为191 mV,并且在电流密度为100 mA·cm−2时,P‑Ov‑CMFO/IF的过电位仅为295 mV,远远优于Ov‑CMFO/IF(388 mV)、CMFO/IF(395 mV)和 P‑Precursor/IF(420 mV),说明氧空位在促进OER活性中起着关键作用,磷原子填充在氧空位中可以稳定氧空位,并且由于磷原子掺杂对催化剂表面电子结构的改变而获得出色的OER活性[24]。含有氧空位的样品Ov‑CMFO/IF比不含氧空位的CMFO/IF性能更好,说明氧空位的引入对材料析氧性能有着一定的促进作用。其余材料P‑Ov‑CLFO/IF、P‑Ov‑CRFO/IF和RuO2在100 mA·cm−2时过电位分别为306、309、438 mV,说明钴的含量对析氧性能也会有影响,适量的钴才能使样品展示出更优异的析氧性能。
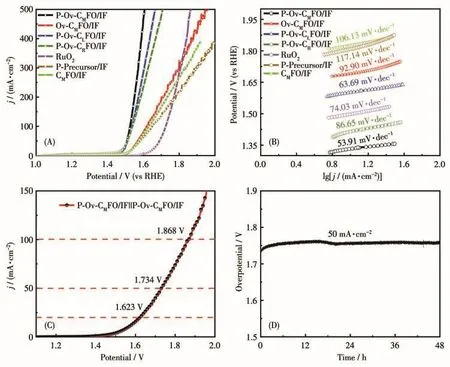
图7 不同样品的(A)LSV曲线和(B)Tafel图;(C)P‑Ov‑CMFO/IF||P‑Ov‑CMFO/IF全解水的j‑V图;(D)P‑Ov‑CMFO/IF全解水连续48 h的稳定性Fig.7 (A)LSV curves and(B)Tafel diagrams of different samples;(C)j‑V plot for overall water splitting by P‑Ov‑CMFO/IF||P‑Ov‑CMFO/IF;(D)Stability of P‑Ov‑CMFO/IF in overall water splitting for 48 h
此外,Tafel斜率也是评估OER动力学的一个重要参数。如图7B所示,P‑Ov‑CMFO/IF的Tafel斜率最低(53.91 mV·dec−1),显示了P‑Ov‑CMFO/IF材料的快速OER动力学。此外,其余6种样品的塔菲尔斜率分别为 92.9 mV·dec−1(Ov‑CMFO/IF)、106.13 mV·dec−1(CMFO/IF)、117.14 mV·dec−1(P ‑Precursor/IF)、74.03 mV·dec−1(RuO2)、63.69 mV·dec−1(P ‑Ov‑CLFO/IF)和 86.65 mV·dec−1(P‑Ov‑CRFO/IF)。综上可知,P‑Ov‑CMFO/IF优于其余6种材料,显示了该催化剂良好的OER动力学特性。可见,P‑Ov‑CMFO/IF表现了出色的HER和OER性能(尤其是在高电流密度下),因此,P‑Ov‑CMFO/IF可以作为一种广泛使用的双功能催化剂。如图7C所示,在一个双电极池(1.0 mol·L−1KOH,pH=14,25 °C)中,在 20、50、100 mA·cm−2下,使用 P‑Ov‑CMFO/IF 获得的电压为 1.623、1.734、1.868 V。如图7D所示,在50 mA·cm−2下,对全解水稳定性进行了测试。P‑Ov‑CMFO/IF||P‑Ov‑CMFO/IF运行48 h,电压变化可忽略不计,说明全解水稳定性较为良好。
3 结论
本研究通过原位浸泡、真空煅烧和磷化处理在泡沫铁基体上制备了纳米花状结构电催化材料,探究了氧空位和磷掺杂对铁钴双金属材料电催化性能的影响。真空处理产生的氧空位,可以加快电子传输速率,稳定形貌,增加活性位点;磷掺杂可以调控CoFe2O4的电子结构,氧空位和磷掺杂的协同作用使得材料表现出极为优异的催化电解水性能。这项工作表明氧空位和磷掺杂可以协同增强铁钴双金属材料电催化性能,为过渡金属基高效电解水催化剂的发展提供了一种新的策略。
猜你喜欢
大学物理(2022年9期)2022-09-28
长江蔬菜(2021年22期)2022-01-12
物理通报(2020年7期)2020-07-01
上海建材(2020年12期)2020-04-13
长江蔬菜(2018年22期)2018-12-25
长江蔬菜(2018年6期)2018-05-08
中国有色金属学报(2018年2期)2018-03-26
中南大学学报(自然科学版)(2016年2期)2017-01-19
中国资源综合利用(2016年7期)2016-02-03
原子与分子物理学报(2015年3期)2015-11-24
