
吡啶盐/咪唑盐调控钴萘基二膦酸配位聚合物的结构和性质
2023-02-27 03:29刘照文王肖阳高文康
无机化学学报 2023年2期
徐 艳 王 宣 刘照文 王肖阳 高文康 崔 磊
(宿迁学院信息工程学院材料科学系,宿迁 223800)
0 Introduction
Coordination polymers(CPs)have received tre‑mendous attention recently due to their versatile struc‑tures and multiple functions that can be designed and tailored.As an important class of inorganic‑organic hybrid materials or CPs,metal phosphonates can show versatile architectures with interesting physical and chemical properties[1‑8].Many metal phosphonates have been constructed by decorating the phosphonate ligand with other coordinating functional groups[9‑13]or intro‑ducing a second auxiliary ligand[14‑17].In principle,a near‑limitless number of metal phosphonates can be obtained through different combinations of metal ions and organic ligands.Therefore,it is crucial to under‑stand the mechanism of structure assembly and the structure‑property relationships of this class of material for the final purpose of designing and synthesizing materials according to on‑demand.Many factors,such as the coordination geometry of the central metal ions,connective modes of the organic ligands,deprotonation of the phosphonic acid group,and synthesis conditions,can affect the final structures.Compared with the car‑boxylate group,the phosphonate group has an addition‑al oxygen atom,featuring one more coordinating site and consequently more coordination modes,which makes it a great challenge to design and synthesize materials with specific structures and functions.Many efforts have been made to understand these by investi‑gating different metal centers,functionalized phospho‑nate ligands,and synthesis conditions.An effective strategy to fabricate metal phosphonates with fascinat‑ing structures is using different templates or mineraliz‑ers in the reaction mixture.A few metal phosphonates are reported based on amine‑templated.Wang Guo‑Ming and co‑workers have taken 1‑hydroxyethane‑1,1‑diphosphonic acid (hedpH4)as the diphosphonate ligand to build a family of open‑framework structures with templated aliphatic amines[16,18‑21].To the best of our knowledge,the syntheses of metal phosphonates using different di‑pyridinium templates in the reaction mixture have not been adopted so far.Here,the naph‑thalene‑diphosphonate ligand(Scheme 1)has been utilized for the generation of more coordination sites,along with di‑pyridinium/imidazolium as template ions for the first time.
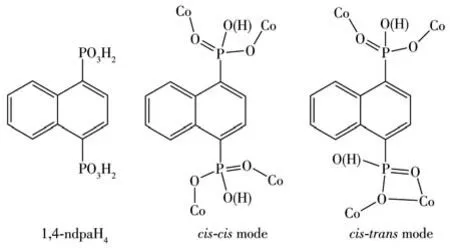
Scheme 1 Molecular structure and coordination modes of 1,4‑ndpaH4ligand
In this work,three cobalt naphthalene‑diphospho‑nates with entirely different structures are obtained under hydrothermal conditions simply by changing the auxiliary ligands and the pH of the reaction mixture.Complexes(1,3‑dppH2)2[Co4(1,4‑ndpa)(1,4‑ndpaH)2(1,4‑ndpaH2)]·6H2O(1)and(1,4‑bixH2)0.5[Co(1,4‑ndpaH)](2)(1,3‑dppH22+=protonated 1,3‑di(4‑pyridyl)propane,1,4‑bixH2+2=protonated bis(imidazol‑1‑ylmethyl)ben‑zene,1,4‑ndpaH4=1,4‑naphthalenediphosphonic acid)show 3D open‑framework structures,respectively.While complex(1,4‑bixH2)0.5[Co2(1,4‑ndpaH)(1,4‑ndpaH2)(H2O)2](3)displays 2D layer structure.Magnet‑ic studies reveal that complexes 1 and 2 show domi‑nant antiferromagnetic interactions.
1 Experimental
1.1 Materials and measurements
1,4‑ndpaH4was synthesized according to the liter‑ature[22].All starting materials were of analytical reagent grade and used as received without further pu‑rification.Elemental analysis for C,H,and N was per‑formed on a Perkin‑Elmer 240C elemental analyzer.Infrared spectra were measured as KBr pellets on a Bruker Tensor 27 spectrometer in 400 ‑4 000 cm−1.Thermogravimetric analysis(TGA)was performed on a METTLER TOLEDO TGA/DSC‑1 over 25‑800 ℃under a nitrogen flow at a heating rate of 10 ℃·min−1.Powder X‑ray diffraction(PXRD)data were collected on a Bruker D8 ADVANCE X‑ray powder diffractome‑ter(Cu Kα,λ=0.154 06 nm)operating at 45 kV and 40 mA over a 2θ range of 5°to 50°at room temperature.The magnetization data were recorded on a vibrating sample magnetometer(VSM)of Quantum Design.The diamagnetic contribution of the sample itself was esti‑mated from Pascal′s constants[23].
1.2 Synthesis
1.2.1 Synthesis of complex 1
A mixture of CoCl2·6H2O(0.476 g,0.1 mmol),1,4‑ndpaH4(0.028 6 g,0.1 mmol),and 1,3‑dpp(0.019 6 g,0.1 mmol)in 10 mL of water,which pH value was adjusted to 4.15 with 0.5 mol·L−1NaOH solution,was sealed in a Teflon‑lined autoclave and heated at 140 ℃for 3 d.After cooling to room temperature,blue rod‑like crystals were collected and washed with water by suction filtration.Yield:55.3 mg.Elemental analysis Calcd.for C66H72Co4N4O30P8(%):C,42.06;H,3.85;N,2.97.Found(%):C,42.11;H,3.81;N,2.83.FT‑IR(KBr,cm−1):3 397(w),3 237(w),1 635(m),1 507(m),1 215(m),1 187(w),1 154(vs),1 123(vs),1 090(vs),1 040(m),1 017(s),963(s),941(s),856(m),812(m),757(m),624(s),570(m),519(s),478(s),439(w),407(w),403(w).
1.2.2 Synthesis of complex 2
A mixture of CoCl2·6H2O(0.047 4 g,0.2 mmol),1,4‑ndpaH4(0.031 2 g,0.1 mmol),and 1,4‑bix(0.025 6 g,0.1 mmol)in 10 mL of water,which pH value was adjusted to 5.6 with 0.5 mol·L−1NaOH solution,was sealed in a Teflon‑lined autoclave and heated at 140 ℃for 3 d.After cooling to room temperature,blue rod‑like crystals were collected and washed with water by suction filtration.Yield:23.8 mg.Elemental analysis Calcd.for C17H15CoN2O6P2(%):C,43.99;H,3.26;N,6.03.Found(%):C,43.83;H,3.29;N,6.12.FT‑IR(KBr,cm−1):3 140(m),3 090(w),1 570(m),1 550(m),1 516(m),1 445(m),1 273(m),1 208(m),1 158(w),1 092(vs),1 016(s),944(vs),863(m),770(s),709(m),619(s),522(s),413(m).
1.2.3 Synthesis of complex 3
Complex 3 was obtained as purple rod‑like crys‑tals by following a similar procedure to that of 2,except that the pH value of the reaction mixture was adjusted to 4.3.Yield:15.9 mg.Elemental analysis Calcd.for C27H27Co2N2O14P4(%):C,38.37;H,3.22;N,3.31.Found(%):C,38.56;H,3.18;N,3.29.FT ‑IR(KBr,cm−1):3 356(s),1 578(m),1 511(m),1 213(s),1 186(vs),1 157(vs),1 086(vs),1 072(vs),1 034(w),1 010(w),970(m),940(vs),756(m),631(s),571(m),524(m),509(m),493(w),436(w).
1.3 Crystallographic data collection and refinement
Single crystals with sizes of 0.15 mm×0.13 mm×0.12 mm for 1,0.12 mm×0.11 mm×0.10 mm for 2,and 0.16 mm×0.15 mm×0.13 mm for 3 were used for struc‑tural determination on a Bruker D8 Venture diffractom‑eter using graphite‑monochromated(Mo Kα,λ =0.071 073 nm)at 100 K.A hemisphere of data was col‑lected in a 2θ range of 3.03°‑58.818°for 1,4.124°‑58.884°for 2,and 2.996°‑58.666°for 3.The numbers of observed and unique reflections are 56 305 and 16 056(Rint=0.031 8)for 1,16 837 and 4 117(Rint=0.029 3)for 2,33 130 and 6 926(Rint=0.036 4)for 3.Using Olex2,the structure was solved with the SHELXT structure solution program using Intrinsic Phasing and refined with the SHELXL refinement package using Least Squares minimization All H atoms were refined isotro‑pically,with the isotropic vibration parameters related to the non‑H atom to which they are bonded.Details of the crystal data and refinements of 1‑3 are summarized in Table 1,and selected bond lengths and angles of 1‑3 are listed in Table S1‑S3(Supporting information).
Table 1 Crystallographic data and structure refinement details for complexes 1⁃3
CCDC:2202596,1;2202597,2;2202598,3.
2 Results and discussion
2.1 Synthesis
Complexes 1‑3 were synthesized under similar experimental conditions except for the auxiliary ligands and the pH value of the reaction mixture(Scheme 2).Pure phases of blue rod crystals of 1 and blue shuttle crystals of 2 were obtained at pH=5.6.When the pH was descended to 4.3,purple rob crystals of 3 were obtained,contaminated with a small amount of 2.We tried to isolate a pure phase of 3 by changing the reaction temperature and solvents but failed.Purple rob crystals of 3 were manually selected under the microscope for subsequent characterization.Its purity was confirmed by the PXRD pattern in compari‑son with that simulated from the single crystal data(Fig.S1‑S3).

Scheme 2 Synthetic routes of complexes 1‑3
2.2 Structure description
Complex 1 crystallizes in the monoclinic system space group P21/c.The asymmetric unit contains four Co(Ⅱ) ions,one 1,4‑ndpa4−ion,two 1,4‑ndpaH3−ions,one 1,4‑ndpaH22−ion,two 1,3 ‑dppH22+ions,and six lattice water molecules(Fig.1a).All Co(Ⅱ)ions have distorted tetrahedral geometry,surrounded by four phosphonate oxygen atoms(O1,O6A,O10B,O21C for Co1,O4,O7,O12D,O13 for Co2;O2E,O16,O18F,O19 for Co3;O8B,O14B,O22,O23G for Co4)(Symme‑try codes:A:−x+1,y−1/2,−z+3/2;B:x−1,y,z;C:x,y−1,z;D:−x+2,y+1/2,−z+3/2;E:x,y+1,z,F:−x+1,−y+2,−z+1;G:−x,−y+1,−z+1).The Co—O bond lengths and O—Co—O angles fall in a range of 0.193 37(15)‑0.19850(15)nmand95.74(7)°‑117.86(6)°,respectively,in agreement with those for the other cobalt phospho‑nates with tetrahedral geometry[24‑25].
The eight phosphonate groups(P1‑P8)connect four Co(Ⅱ) ions using two of its three phosphonate oxy‑gen atoms in a cis‑cis coordination mode(Scheme 1),forming an infinite chain.Notably,P2,P3,P4,and P6 are singly protonated,while the remaining phosphonate groups are all fully deprotonated.The{CoO4}polyhe‑dra are each involved in the corner sharing with four{PO3C}tetrahedrons,forming a 1D inorganic chain in the ac plane(Fig.1b).The Co…Co distances over the double O—P—O bridges are in a range of 0.417 71(7)‑0.454 40(8)nm.The inorganic chains are further cross‑linked by naphthalene groups,leading to a 3D open‑framework structure(Fig.1c).Notably,in the 3D struc‑ture,channels extend indefinitely along the a‑axis.The protonated 1,3‑dppH22+occupies these channels along with free water molecules(Fig.1d),interacting with each other and the framework through hydrogen bonds(N1…O1W:0.270 3(2)nm;N2…O5W:0.270 4(3)nm,N3…O4W:0.272 9(3)nm;N4…O6W:0.278 64(3)nm)(Table S2).
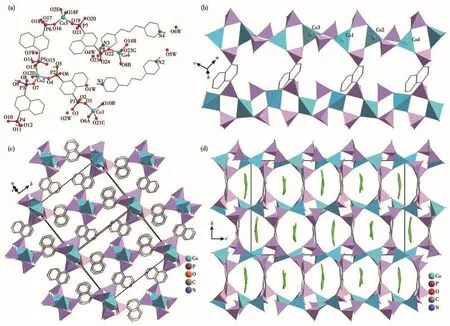
Fig.1 (a)Building unit of 1 with atomic labeling scheme;(b)1D inorganic chain;(c)Inorganic chains cross‑linked by naphthalene groups,where 1,3‑dppH2 2+ions are omitted for clarity;(d)View of the supramolecular structure of 1 along the[011]direction showing the template,1,3‑dppH2 2+,placed in the channel
Complex 2 crystallizes in the monoclinic system space group P21/n.The asymmetric unit contains one Co(Ⅱ) ion,one 1,4‑ndpaH3−ion,and half a 1,4‑bixH2+2ion(Fig.2a).Like 1,Co(Ⅱ) ion in complex 2 also dis‑plays a distorted tetrahedral geometry,in which all of the four coordination sites are occupied by phospho‑nate oxygens(O1,O2A,O4,and O6B)(Symmetry codes:A:1−x,−y,1−z;B:0.5+x,0.5−y,0.5+z)from four equivalent 1,4 ‑ndpaH3−ions.The Co—O bond lengths are 0.193 06(16)‑0.195 63(15)nm,and O—Co—O angles are 105.52(7)°‑116.99(6)°.Like those in complex 1,the phosphonate ligands in 2 adopt a cis‑cis coordination mode (Scheme 1),behaving as a quadrdentate ligand,and coordinating with four Co(Ⅱ)ions.Thus each{CoO4}tetrahedron is corner‑shared with four{PO3C},forming an infinite chain along the a‑axis(Fig.2b).The Co…Co distances over the double O—P—O bridges are 0.460 97(4)and 0.456 61(4)nm.Like those found in complex 1,the chains are also linked by naphthalene groups,forming a 3D supramo‑lecular structure.Furthermore,the protonated template of 1,4‑bixH22+is suspended in the skeletal voids of the crystal structure.Hydrogen bonds exist between the phosphonate oxygen atoms and 1,4‑bixH22+counterions(N1…O5:0.273 4(3)nm)along the b‑axis(Fig.2c).
Complex 3 crystallizes in the monoclinic system space group P21/n and shows a 2D layer structure.The asymmetric unit contains two Co(Ⅱ) ions,one 1,4‑ndpaH3−ion,one 1,4 ‑ndpaH22−ion,two coordination water molecules,and half a 1,4‑bixH22+ion.Compared with complex 2,Co1 is five‑coordinate with a distorted trigonal‑bipyramidal geometry in complex 3,in which four of the five coordination sites are occupied by phos‑phonate oxygens(O1,O7,O5A,O12B,Symmetry codes:A:3/2−x,−1/2+y,1/2−z;B:1/2−x,−1/2+y,1/2−z)from four equivalent 1,4‑ndpaH3−ions and the remain‑ing one is filled with the oxygen atom(O13)of the coor‑dination water molecule(Fig.2d).Co2 has a distorted octahedral environment with the five sites occupied by five phosphonate oxygen(O1C,O2C,O4,O9D,O10,Symmetry codes:C:3/2−x,1/2+y,1/2−z;D:1/2−x,1/2+y,1/2−z),and one coordination water atom(O14).The Co—O bond lengths are between 0.199 02(13)and 0.226 70(14)nm and the O—Co—O bond angles lie in a range of 84.68(5)°‑164.51(5)°(Table S4).Two tetra‑dentate naphthalene phosphonate ligands differ slightly in their protonation,monoprotonated(1,4‑ndpaH3−)and bi‑protonated(1,4‑ndpaH22−).Unlike those in complexes 1 and 2,the phosphonate ligands in 3 adopt a cis‑trans coordination mode(Scheme 1).They chelate and bridge four cobalt ions.Each{Co1O5}polyhedron is corner‑shared with four{PO3C}tetrahedrons,while the{Co2O6}polyhedron is involved in corner‑sharing with three{PO3C}tetrahedrons and edge‑sharing with one{PO3C}tetrahedron.Therefore,the Co1 and Co2 are bridged by one μ2‑O(P)and one O—P—O linker,form‑ing a dimeric unit of Co2.The Co1…Co2 distance with‑in the dimer is 0.388 94(4)nm.The equivalent dimers are connected by two{PO3C}tetrahedrons to form an infinite chain running along the a‑axis(Fig.2e).The distance between the dimers is 0.415 04(4)nm.The chains are cross‑linked by naphthalene groups,forming a 2D wave layer in the ab plane(Fig.2e).The protonat‑ed 1,4‑bixH22+are filled between layers(Fig.2f).

Fig.2 (a)Building unit of 2 with the atomic labeling scheme;(b)Inorganic chains cross‑linked by naphthalene groups;(c)3D polyhedral view of complex 2;(d)Building unit of 3 with the atomic labeling scheme;(e)Wave single layer structure of 3 where the inorganic chains are cross‑linked by naphthalene;(f)Packing diagram of 2 in ABAB mode viewed along the a‑axis
All the complexes exhibit di‑pyridinium/imidazoli‑um templated 3D or 2D extended structures on the connectivity between the Co2+ions and naphthalene‑diphosphonate units.The reaction conditions in all the cases are similar,but the observed structural differenc‑es are mainly due to the presence of variable dipyri‑dine molecules and pH.Both complexes 1 and 2 have analogous 3D open‑framework structures connecting through metal and diphosphonates (naphthalene diphosphonic acid),while the di‑pyridinium/imidazoli‑um template is different in the framework.In the struc‑ture of 1,the template 1,3‑dppH22+is placed in the channel formed from{CoO4}tetrahedron and{CPO3}tetrahedron connected by naphthalene groups(Fig.1d)with a channel size of 0.64 nm×0.64 nm(shortest atom‑atom contact distances,not including the van der Waals radii).The protonated 1,3‑dppH22+molecules occupy these channels along with free water molecules and interact with each other and with the framework through hydrogen bonds.The above channel is growing along the a‑axis,which is different from the direction of the inorganic chain.In the structure of 2,templated 1,4‑bixH22+cations are situated inside the bigger chan‑nel of size(1.34 nm×0.66 nm),constructed from four inorganic chains made up of{CoO4}tetrahedron and{PO3C}tetrahedron along the a‑axis(Fig.2c).The pro‑tonated 1,4‑bixH22+are stabilized through extensive N—H…O hydrogen bonding interactions with the framework.Complexes 2 and 3,despite slight distinc‑tion of pH in the reactions,the same template effect leads to different topological structures.Comparing with complex 2,complex 3 exhibits an anionic layer with protonated di‑pyridinium/imidazolium template located in interlayer space.Since at a lower pH value(4.3),there are diprotonated diphosphonates(1,4‑ndpaH22−),which are absent in 1 and 2.It is worth men‑tioning that although cobalt phosphonates with open‑framework structures composed of inorganic chains and organoamine ‑directed were previously reported[21,26‑27],none of them contain a di‑pyridinium/imidazolium template.
2.3 Thermal stability of the complexes
The TGA curves for complexes 1‑3 are shown in Fig.3.The TGA curve of complex 1 revealed a multi‑step weight loss process.The first step below 175℃corresponds to a weight loss of 5.47%,attributed to the release of six lattice water molecules(Calcd.5.73%).The dehydrated samples were stable up to 330℃,above which a second weight‑loss step was observed with the removal of two 1,3‑dppH22+ions(19.18%)(Calcd.19.76%).The third step was observed above 525℃,corresponding to the decomposition of the organic ligands and the collapse of the structure.Ther‑mal analysis revealed that complex 2 was stable up to 400℃,above which the curve drops rapidly,due to the burn of the organic components and the collapse of the 3D structure.For complex 3,the first step occurred at about 240℃with a weight loss of 4.46%,in agreement with the removal of two lattice water molecules(Calcd.4.26%).This was followed by a short plateau until 355℃,above which a quick weight loss was observed corresponding to the release of the uncoordinated 1,4‑bixH22+ions and the decomposition of the organic components.
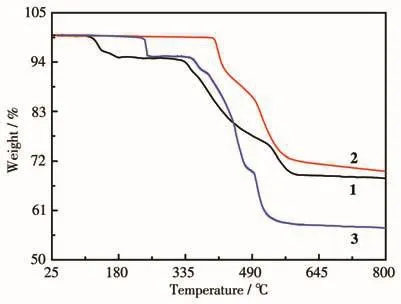
Fig.3 TGA curves for complexes 1,2,and 3
2.4 Magnetic properties of complexes 1 and 2
We attempted to synthesize sufficient amounts of complex 3 to characterize its magnetic properties but unfortunately failed.The temperature‑dependent magnetic susceptibilities of 1 and 2 were measured in a temperature range of 2‑300 K under an applied field of 1 kOe(Fig.4).The χMT values for each Co(Ⅱ) at 300 K were 2.36 cm3·mol−1·K for 1 and 2.41 cm3·mol−1·K for 2,and both are larger than the spin‑only value of 1.875 cm3·mol−1·K for one spin ‑only Co (Ⅱ) (S=3/2,g=2).Since the ground state of a tetrahedral Co(Ⅱ)is4A2,the higher value of χMT could be attributed to the orbital contribution from the lowing excited states[25].Upon cooling,the χMT products of 1 and 2 gradually decreased to 0.35 and 0.29 cm3·K·mol−1at 2 K,respectively.Above 100 K,the susceptibility data follow the Curie‑Weiss law with the Curie constants(C)and Weiss constants(θ)of 2.77 cm3·K·mol−1and−20.98 K for 1 and 2.45 cm3·K·mol−1and −4.59 K for 2,respectively.The negative Weiss constant is attribut‑ed to the antiferromagnetic exchange couplings between the Co(Ⅱ) centers and/or the spin‑orbital coupling of the single Co(Ⅱ)ion.For 2,the χMvs T plot shows a peak at 6 K confirming the presence of antiferromagnetic interactions between the Co(Ⅱ)centers.
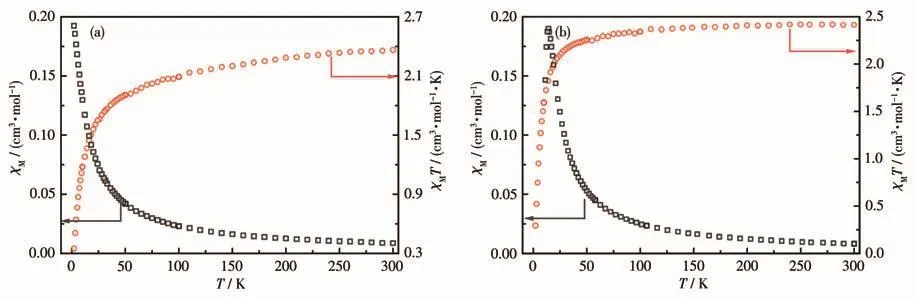
Fig.4 χMand χMT vs T plots under at 1 kOe dc field for 1(a)and 2(b)
3 Conclusions
In summary,we report for the first time that di‑pyridinium/imidazolium templated modulated struc‑ture in metal phosphonates.Three new cobalt naphtha‑lene‑diphosphonates,namely(1,3‑dppH2)2[Co4(1,4‑ndpa)(1,4‑ndpaH)2(1,4‑ndpaH2)]·6H2O(1),(1,4‑bixH2)0.5[Co(1,4‑ndpaH)](2),and(1,4‑bixH2)0.5[Co2(1,4‑ndpaH)(1,4‑ndpaH2)(H2O)2](3),were successfully prepared by the hydrothermal method in the reaction of CoCl2·6H2O with 1,4‑ndpaH4and 1,3‑dpp for 1 and 1,4‑bix for 2,3 at different pH values.Complexes 1 and 2 have 3D open‑framework structures,constructed by inorgan‑ic chains cross‑linked by naphthalene groups,while complex 3 exhibits a 2D layer structure,constructed by inorganic chains connected by naphthalene groups.The protonated di‑pyridinium/imidazolium templates,1,3‑dppH22+for 1,1,4‑bixH22+for 2 and 3,fill and com‑pensate the negative charge.Magnetic studies reveal that dominant antiferromagnetic interactions between the magnetic centers are propagated in complexes 1 and 2.The present examples are not only enriching the field of di‑pyridinium/imidazolium ‑templated open‑framework materials but also open possibilities for investigations of new phosphonates using different templates and metal combinations.
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
中国机械工程(2022年22期)2022-11-25
中国机械工程(2022年7期)2022-04-20
佛山科学技术学院学报(自然科学版)(2022年1期)2022-02-15
华人时刊(2022年17期)2022-02-15
今日农业(2020年23期)2020-12-15
——材料科学与工程
湖南人文科技学院学报(2020年3期)2020-06-08
中国机械工程(2019年17期)2019-09-19
中国机械工程(2018年21期)2018-11-13
中国机械工程(2018年4期)2018-03-06
中国医院院长(2017年21期)2018-01-19
