
间变性大细胞激酶阴性CD30阳性原发性皮肤间变性大细胞淋巴瘤7例临床分析
2023-02-27 05:56关军程平王兰兰帅华洲胡彬张婷陈柳青程辉邹亮
临床内科杂志 2023年2期
关军 程平 王兰兰 帅华洲 胡彬 张婷 陈柳青 程辉 邹亮
原发性皮肤淋巴瘤(PCL)作为淋巴瘤分类中独立的一类,具有不同于其他淋巴瘤的特异性临床表现和组织病理学特征。其中,原发性皮肤CD30阳性(CD30+)T细胞淋巴增殖性疾病包括淋巴瘤样丘疹病和原发性皮肤间变性大细胞淋巴瘤(PC-ALCL),后者约占皮肤T细胞淋巴瘤的0.9%[1]。本研究回顾性分析了我院血液内科诊治的7例间变性大细胞激酶(ALK)阴性(ALK-)CD30+PC-ALCL患者的临床特征,现报告如下。
对象与方法
1.对象:回顾性纳入2016年1月~2020年10月在我院血液内科诊治的7例ALK-CD30+PC-ALCL患者,所有患者均经组织学及影像学检查确诊为CD30+PC-ALCL。
2.方法
(1)临床资料和临床表现收集:临床资料包括性别、年龄、发病时间、EB病毒(EBV)-DNA、病灶范围、治疗方案、疗效及结局、存活情况、肿瘤病理组织学检查结果及T细胞抗原受体(TCR)基因重排检测结果。临床表现包括发病部位、病灶情况、骨髓浸润情况、皮肤病灶范围、合并EBV感染情况、治疗方案、正电子发射计算机断层显像(PET)-CT或增强CT检查结果、淋巴结活检或淋巴结穿刺活检结果、随访预后情况。根据皮肤病灶范围分为孤立型、区域型(伴或不伴局部淋巴结转移)、广泛型,其中孤立型定义为仅有1个病灶,区域型定义为多个病灶局限于1个解剖区域或2个连续解剖区域,广泛型定义为多个病灶累及2个不连续或3个及以上解剖区域。治疗方案包括手术切除、放疗及化疗。化疗方案主要为CHOP方案(环磷酰胺、多柔比星、长春新碱、泼尼松)、小剂量甲氨蝶呤(MTX)方案(MTX 10 mg/m2口服,每周1次);维甲酸20 mg口服,每天1次,部分患者联用依托泊苷。疗效评估标准参照文献[1]。
(2)随访:以2021年6月30日为随访截止日。随访过程中记录患者的病灶变化,评估缓解情况,若患者再发肿块、结节,再次行肿块活检组织病理学检查,并通过胸腹部CT、淋巴结彩色超声、包块彩色超声、骨髓穿刺检查等重新评估系统受累情况。
3.统计学方法:采用SPSS 23.0软件进行统计分析。不符合正态分布的计量资料以M(P25,P75)表示;计数资料以例数表示。
结 果
1.临床资料:7例患者中男2例,女5例,年龄29~72岁,中位年龄62(29,72)岁,4例接受系统化疗,2例接受局部放疗,1例拒绝治疗。患者的临床资料见表1。截止随访日期,6例存活,其中2例无病生存,4例带病生存;1例死亡。所有患者肿瘤细胞表面均弥漫性表达CD30,每例患者病理切片中CD30阳性表达率均大于90%,同时表达白细胞共同抗原(LCA),部分患者表达颗粒酶B、T细胞胞浆内抗原(TIA-1),不表达CD56、上皮细胞膜抗原(EMA)、CD20、EBV编码的小分子量RNA(EBER)。所有患者肿瘤细胞表面细胞周期调节蛋白-67(Ki-67)阳性表达率均大于50%。见图1。所有患者组织病理切片TCR基因重排检测结果均为阳性。
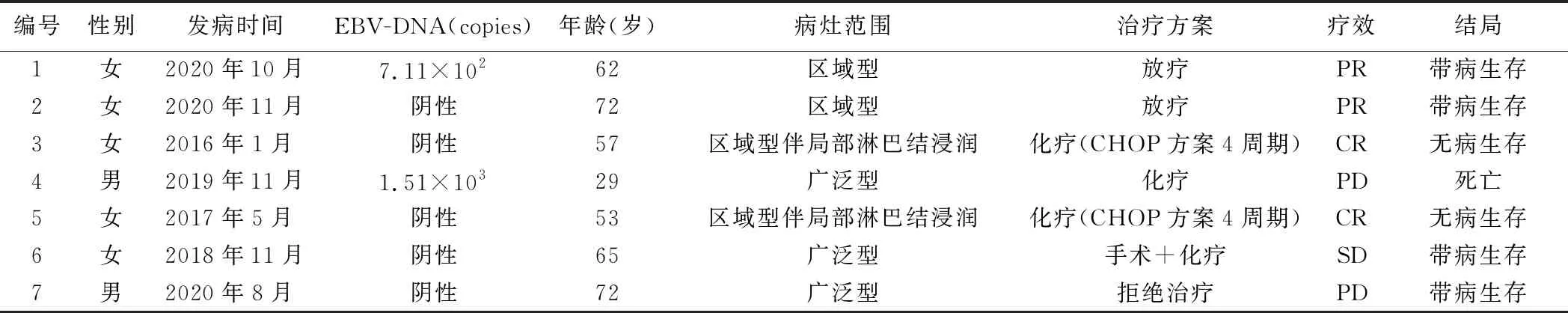
表1 7例患者的临床资料
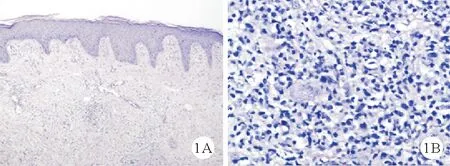
图1 病例6肿瘤组织病理切片可见弥漫单一核细胞浸润,部分细胞体积大,胞浆淡染,核深染伴较多嗜酸性粒细胞(苏木素-伊红染色;A:×40;B:×200)
2.临床表现:患者发病部位主要集中在头面部、四肢、腹部,病灶在初诊时表现为结节或肿块,其中发生于上肢2例、下肢2例、头面部2例、腹部1例。所有患者均无骨髓浸润,其中4例患者形成局部凹陷性溃疡,迁延不愈。2例患者合并EBV感染。病例1、2皮肤病灶呈区域型(图2A、2B),行局部放疗。病例3、5皮肤病灶呈区域型,局部溃疡并继发感染,存在局部淋巴结肿大(直径>1 cm),PET-CT结果提示局部淋巴结标准摄取值(SUV)轻度增高,行淋巴结活检或淋巴结穿刺活检,病理学检查未发现肿瘤细胞;这两例患者接受CHOP方案化疗4周期后随访至今,淋巴瘤无复发表现,溃疡面在局部换药及系统治疗1个月后均愈合。病例4于阑尾炎手术后反复出现腹部切口皮肤软组织感染,多次病理检查未发现淋巴瘤病变,后于2019年12月腹部切口出现包块,病理检查示ALK-ALCL,患者因疫情原因未按化疗周期规律化疗,其皮肤病变情况见图3;该患者后于外院接受CHOP、ESHAP(依托泊苷联合环磷酰胺、多柔比星、长春新碱、地塞米松)方案化疗,皮肤包块仍进展迅速,于2020年5月至我科就诊,先后行西达本胺联合MTX治疗,拟行嵌合抗原受体T(CAR-T)细胞治疗,但患者未及时治疗,继发腹腔感染死亡。病例6呈孤立性病灶,于2018年接受下肢局部病灶切除治疗,两年后于同侧及对侧肢体再发多处肿块,再次取活检,病理结果证实仍为ALCL;先后行CHOP方案、小剂量MTX治疗、依托泊苷治疗效果均不佳,病灶短期缩小后再次进展,反复合并皮肤软组织感染,目前予维甲酸联合GDP方案(吉西他滨、顺铂、地塞米松)治疗(图2C)。病例7病灶局限于头面部,患者及其家属拒绝静脉化疗及放疗,原发病灶逐渐增大(图2D)。
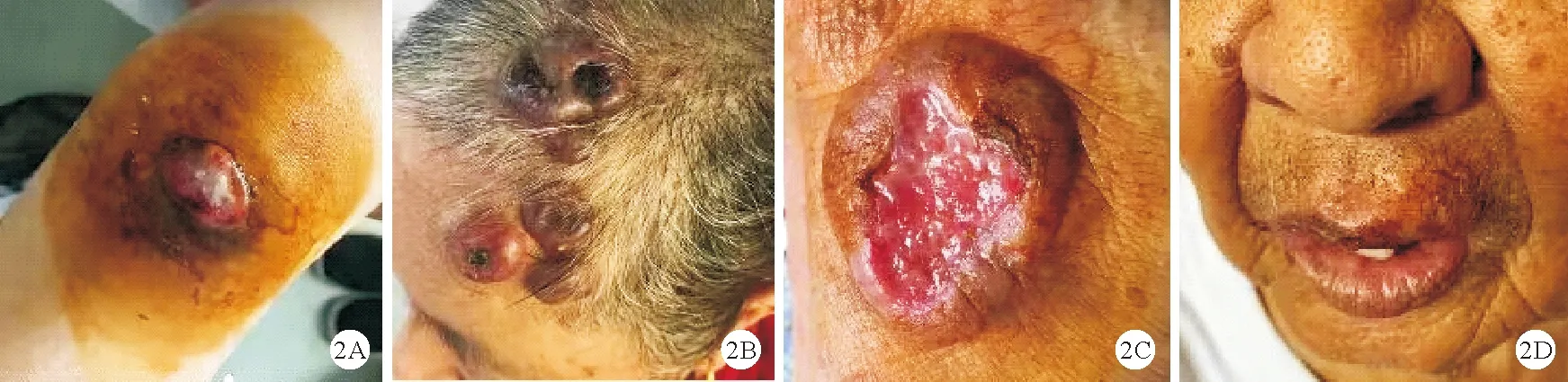
图2 病灶大体形态图(A:病例1;B:病例2;C:病例6;D:病例7)
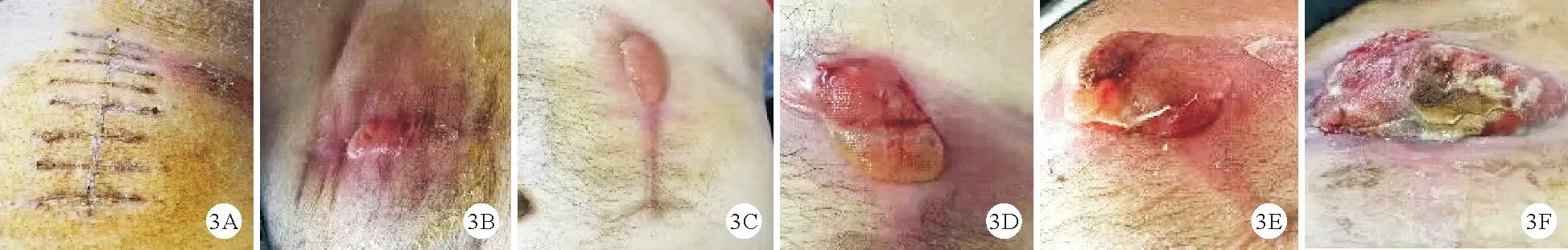
图3 病例4病灶大体形态变化趋势图(前期中止化疗后,患者腹部病灶从A局部手术切口发展至B中的局部隆起物,C、D、E可见病灶范围逐渐增大,并出现红肿、渗液、脓性分泌物,多次分泌物培养显示金黄色葡萄球菌阳性,最终发展至F中的7 cm×5 cm包块,伴局部溃疡形成,软组织感染)
3.预后分析:除1例患者拒绝治疗外,其余均接受治疗,治疗总反应率为83.3%。根据病灶范围,区域型2例、区域型伴局部淋巴结浸润2例、广泛型3例。对区域型病灶伴局部淋巴结浸润患者采用系统化疗,未出现化疗相关严重并发症,治疗后均达CR,无病生存期达4年以上;区域型病灶患者通过局部放疗带病生存;广泛型患者对化疗不敏感,尽管给予系统化疗,原发病仍控制不佳,1例死亡,2例带病生存。4例伴顽固性溃疡合并软组织感染患者反复换药、抗感染治疗无效,其中3例接受系统化疗后溃疡逐渐愈合。
讨 论
PC-ALCL是一种由间变性、多形性或免疫母细胞形态的非典型大淋巴细胞形成的肿瘤,多数肿瘤细胞(>75%)表达CD30[1]。ALK在PC-ALCL中往往表现为阴性,与系统性ALCL相比,其临床表现偏惰性,也有少部分患者表现为ALK阳性。大部分肿瘤细胞不表达或低表达B淋巴细胞瘤-2(BCL-2),由于蕈样肉芽肿(MF)可能进展为原发皮肤大细胞性T细胞淋巴瘤,故在诊断PC-ALCL时需排除MF,并注意有无人类免疫缺陷病毒(HIV)感染、器官移植等病史[2]。
PC-ALCL临床常表现为皮肤孤立的红色或紫红色结节或肿物,直径1~10 cm不等,晚期多表现为溃疡性损害,中央坏死,边缘呈堤状隆起,约20%的患者可表现为多发性皮损,25%~31%的患者皮损可部分或完全自行消退,但会在皮肤以外器官复发[3-4]。皮肤以外的扩散罕见,约10%的患者可出现局部淋巴结浸润[3]。PC-ALCL可能会合并异位性皮炎、虫咬后高敏反应、嗜血细胞综合征、伴抗利尿激素异常分泌的副肿瘤综合征等,部分PC-ALCL的发生甚至可能与药物相关[5]。本研究结果发现,患者一旦形成溃疡性损害,往往呈现为顽固性溃疡,易反复感染,不易愈合。
迄今为止,PC-ALCL的细胞学起源尚未明,其在分子生物学上多表现为ALK阴性、双特异性磷酸酶22(DUSP22)阴性及肿廇蛋白P63(TP63)阴性,并伴杀伤细胞免疫球蛋白样受体3DL2(KIR3DL2)强表达,部分患者可具有表观遗传相关基因异常,如Zeste基因增强子同源物2(EZH2)过表达等[5]。CD30-CD30L通路可能参与PC-ALCL发展过程[6]。少数患者(<10%)伴核仁磷酸蛋白(NPM1)-ALK,部分患者检出NPM1-酪氨酸激酶2(NPM1-TYK2)融合蛋白。6p25.3重排、肿瘤坏死因子受体相关因子1与PC-ALCL密切相关,涉及的基因包括干扰素调节因子4(IRF4)、DUPS22等,伴有IRF4重排的患者更多见淋巴结浸润,伴DUPS22阳性表达的患者则更易表现出亲表皮的小淋巴细胞浸润[7-8]。该类淋巴瘤常局限于皮肤,可能与其细胞表面趋化因子C-C-基元受体(CCR)10和CCR8表达有关。在PC-ALCL中,角化细胞表面Toll样细胞受体(TLR)如TLR-2、TLR-4和TLR-7等均有不同程度的上调表达,这种现象可能与感染性因素有关[9-10]。肿瘤坏死因子α(TNF-α)、肿瘤坏死因子相关的凋亡配体、CD95配体、转化生长因子(TGF)-β/SMAD、Jun B原癌基因(JUNB)、信号传导转录激活因子5A(STAT5A)、类视黄醇X受体α(RXRA)、IL-2受体α(IL-2Rα)等途径可能参与PC-ALCL的病变发展,其中,JUNB与CD30过表达存在一定联系[11-12]。5-羟甲基胞嘧啶(5hmC)表达缺失在CD30+淋巴细胞增殖性疾病中具有一定的特异性,并能反映DNA甲基化效应,有助于鉴别反应性或炎症性CD30+淋巴细胞增多[13]。
PC-ALCL患者预后主要与病灶范围有关。初始局部治疗后,约30%~64%的患者可能会复发,中位无病生存时间约为16个月,复发部位主要包括其他皮肤区域、局部淋巴结[3,14]。相关研究结果显示,局部皮肤组织复发、局部淋巴结转移、Ki-67水平与患者不良预后无明显关联[3-4,15]。目前,腿型或上肢的广泛型病变是经临床证实对PC-ALCL具有统计学意义的独立预后不良因素[5]。本研究的病例中,区域型的患者上肢PC-ALCL(病例1、2)治疗效果较好,远期预后良好,这可能与其接受系统化疗有关。
对PC-ALCL患者,采用何种治疗手段需要综合评估。不同临床表现的患者,倾向采用不同的治疗策略。手术切除、放疗在治疗孤立性或区域性PC-ALCL中表现出较高的短期缓解率,但复发率达43%~51%;广泛型PC-ALCL可采用多药化疗,CR率可达92%,但复发率达62%[1,3]。本研究中,病例3及病例5伴有淋巴结浸润,接受系统化疗后预后良好,未出现不良反应,其中病例3病灶图片见于本中心既往报道[15];病例6起病时表现为右下肢局部包块,接受手术切除治疗,两年后对侧下肢再发多个包块,由于包块较大,未行手术治疗,虽然接受化疗,但仍无法缓解,合并顽固性溃疡创面及软组织感染,联合放疗后疾病才得以控制。病例5为广泛型,其表现为高度侵袭性,对多种化疗药物不敏感,后期包块生长迅速,局部皮肤组织溃疡、感染,肿块压迫、浸润肠道,失去手术切除时机。我们考虑对存在大包块、淋巴结转移的患者,尤其是完整手术切除较为困难的患者,早期系统化疗可能有助于争取治疗时机;而对于对初始治疗不敏感、疾病评估为SD或PD的患者,建议尽早评估新药治疗或临床试验的利弊与可行性。接受放疗的区域型病灶患者,尽管病灶大小稳定,但其溃疡、包块长期存在,影响生活质量及外观。结合本中心的治疗体会,尽管文献报道PC-ALCL患者预后较好,但对于合并病灶部位反复感染、溃疡的患者来说,其生活质量大大下降,即使手术、放疗可缓解一定症状,但顽固性溃疡、软组织感染可能会持续存在,因此建议对该类患者采取更为积极的系统治疗及治疗策略。
PC-ALCL其他治疗方式还包括异维A酸、贝沙罗汀、干扰素、沙利度胺、类固醇、激光、局部热疗及造血干细胞移植等[15]。CD30单克隆抗体等新型治疗药物在PC-ALCL中的应用值得进一步研究。Brentuximab vedotin(BV)在系统性ALCL中的疗效较为明确,其在PC-ALCL中的应用较少。Enos等[16]通过系统回顾分析7例患者使用BV治疗PC-ALCL,结果显示总反应率和CR率均为100%,平均缓解持续时间为7.6个月。一项关于BV治疗皮肤T细胞淋巴瘤的国际多中心研究中,接受治疗的PC-ALCL患者中位缓解时间约为7.6个月[17]。
尽管本中心的患者有2例为EBV-DNA阳性,EBV与淋巴瘤患者预后有着密切联系,但在PC-ALCL中,很少有肿瘤细胞表达EBV相关抗原,若患者肿瘤细胞出现EBER阳性,需要排查同时伴CD30和T细胞标记的B细胞淋巴瘤,如浆母细胞淋巴瘤、原发性渗出性淋巴瘤[18-19]。本研究中的7例患者EBER检测结果均为阴性,且EB感染与患者预后无明显关联。
由于PC-ALCL尚缺乏大数据分析,尽管大部分患者预后良好,但在如何进一步预防复发、改善生活质量方面仍需探索最佳治疗策略。本研究结果显示,对于区域型、广泛型患者应采取不同的治疗手段,生活质量、外观等指标可能也需要作为是否行化疗的评估因素。针对区域型病灶患者,早期化疗相比放疗或观察等待是否获益更大、广泛型患者是否需要早期启动新药治疗均需进一步研究。
猜你喜欢
传染病信息(2022年3期)2022-07-15
中学生数理化·七年级数学人教版(2022年11期)2022-02-22
天津医科大学学报(2021年4期)2021-08-21
数学物理学报(2021年2期)2021-06-09
养生保健指南(2019年11期)2019-12-17
发明与创新(2016年38期)2016-08-22
艺术生活-福州大学厦门工艺美术学院学报(2016年3期)2016-07-31
中华老年多器官疾病杂志(2016年9期)2016-04-28
磁共振成像(2015年5期)2015-12-23
天津医科大学学报(2015年3期)2015-06-05
