
SON 基因异常致ZTTK 综合征3 例临床及遗传学分析
2023-02-27 04:28赵培伟黄玉凤何学莲朱红敏
临床儿科杂志 2023年2期
赵培伟 毕 博 张 蕾 黄玉凤 谭 黎 何学莲 朱红敏
华中科技大学同济医学院附属武汉儿童医院(湖北武汉 430016)
ZTTK 综合征(Zhu-Tokita-Takenouchi-Kim syndrome,ZTTK,MIM#617140)是一种罕见的常染色体显性遗传的累及多系统异常的遗传病,Kim 等于2015 年首次报道,其发病率不详[1-2]。该疾病的主要临床表现为发育落后、智力低下、面容异常、肌张力异常、骨骼畸形以及头颅MRI 表现异常等[3]。研究表明SON基因功能丧失变异(LOF)是导致该疾病发生的主要原因[4]。截止到目前,国外期刊及Decipher等数据库共收录了61例ZTTK综合征患者,国内罕见报道。本研究分析利用全外显子测序技术(WES)诊断的3 例ZTTK 综合征患儿,探讨该类疾病的临床表现以及遗传学特点,以加深临床医师对该病的认识。
1 对象与方法
1.1 研究对象
本研究纳入2019 年6 月至2021 年12 月来武汉儿童医院就诊的3例生长发育迟缓及智力落后的患儿,男2 例,女1 例,年龄10 个月~3 岁8 个月(中位年龄13 个月)。本研究得到患者家属知情同意以及医院伦理委员会的批准。
1.2 全外显子测序分析
采集患儿及其家属外周血2 mL,采用天根生物公司外周血基因组DNA 提取试剂盒提取患者DNA 并测定浓度,由第三方检测公司(北京全普基因)进行全外显子测序,测序的原始数据与参考基因组比对,然后采用GATK 以及VarScan 软件进行变异注释分析,利用Annovar 软件及数据库评定变异位点的生物学影响;采用Sanger测序技术验证患者及家属的变异位点,并用ACMG 指南评估其临床意义。
2 结果
2.1 患者临床资料
3 例患儿,男2 例,女1 例,年龄10 个月~3 岁8个月(中位年龄13 个月),临床表型为面容异常、发育迟缓、智力低下、小头畸形、肌张力异常;手关节异常或足外翻,肾盂肾炎、隐睾以及颅缝早闭等症状。其中2例患儿头颅MRI可见脑沟增宽、脑发育不良等异常,患儿1存在听力异常等症状,见表1。

表1 3例ZTTK综合征患儿的临床表现
2.2 遗传学分析结果
测序原始数据经过生物信息学分析注释以后结合患儿的家族史、临床表型发现3 例患儿均存在SON基因(NM_138927)变异,患儿1 存在c.3020G>A(p.R 1007 H)杂合变异,该氨基酸位点在不同的物种之间保守,见图1,且文献及数据库中未见收录,患儿父母未见异常;根据ACMG 指南该位点为可能致病(PS2+PM2+PM)。患儿2存在c.1195delG(p.V 399 fsTer 1)移码变异,其父母未见异常,文献及数据库未见报道,根据ACMG 指南,该位点为致病性变异(PVS 1+PS 2+PM 2)。患儿3 存在c.5753_5756delTTAG(p.V1918EfsTer87)移码变异,其父母未见异常,见图1;根据ACMG指南,该位点为致病性变异(PVS1+PS2+PM2)。3例患儿均诊断为SON基因异常导致的ZTTK综合征。
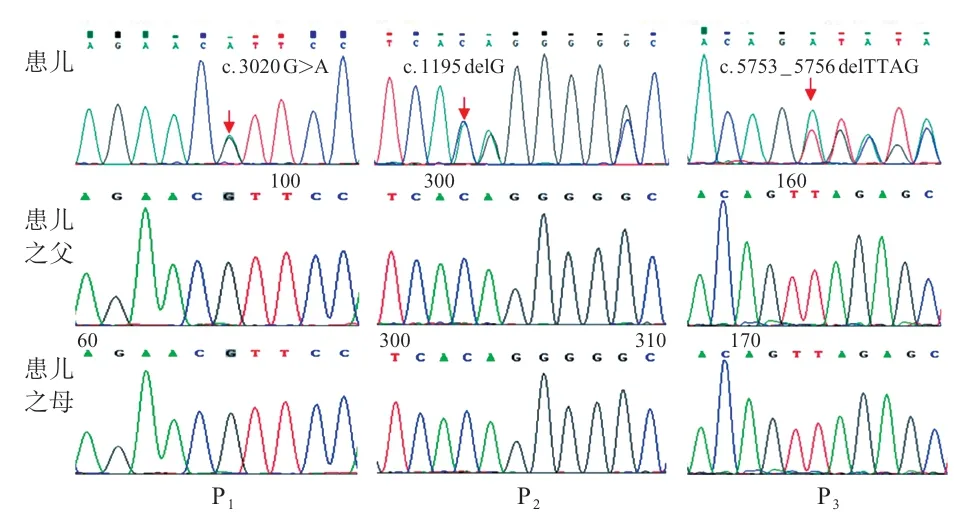
图1 患儿及父母SON 者遗传学检测结果
3 讨论
SON基因(NM_138927.2)定位于21号染色体21q22.11区域,含有12个外显子,编码含有2 426个氨基酸的蛋白,该蛋白含有一个富含精氨酸/丝氨酸功能区(R/S rich domain),一个G-patch功能区以及一个双链RNA集合功能域[5]。SON蛋白是一个DNA或RNA结合蛋白,作为mRNA剪接加工复合物成员之一,SON异常可影响RNA的异常剪接以及基因的表达等[6]。文献及数据库中收录SON基因异常可导致ZTTK综合征(OMIM 617140),其主要的临床表现为发育落后智力低下、肌张力异常、面容异常、骨骼畸形等。在人类细胞中敲除SON后发现细胞迁移相关基因、代谢相关基因以及线粒体功能相关基因等均发生了显著的改变[7-8]。模式动物研究发现,在斑马鱼中敲除SON基因后,斑马鱼表现出发育迟缓、骨骼畸形等症状与ZTTK 综合征表型相似[1];在小鼠模型中敲除SON亦有ZTTK相似的表现[9]。
本研究中的3 例患儿临床表现为面容异常、发育落后、智力低下、小头畸形、肌张力异常;手关节异常或足外翻,肾盂肾炎、隐睾以及颅缝早闭等症状。其中两例(P 1 和P 3)患儿头颅MRI 可见脑沟增宽、脑发育不良等异常;此外例1 存在听力异常。遗传学检测发现3 例患儿均存在SON基因变异,例1 存在错义变异,例2 和3 存在移码变异,结合ACMG 指南结合患者的临床表现,3 例患儿诊断为SON基因异常导致的ZTTK 综合征。目前ZTTK综合征患者尚无有效的治疗方法,临床均采取对症治疗,比如语言康复训练、骨骼校正以及抗癫痫治疗等。
以“SON基因”以及“SONgene”为关键词检索中国知网、万方医学数据库、PubMed、Decipher、HGMD 等数据库,其中国内数据库未见ZTTK 综合征患者报道,PubMed 数据库共检索到到51 例ZTTK 综合征患者,中文期刊数据库检索到1 例患者[1-4,10-15],Decipher 数据库收录9 例患者。其主要的临床表现依次为发育迟缓及智力低下(100%)、面容异常(93.3%)、头颅MRI 异常(84.3%)以及66.7%的患者存在眼睛或视力的异常;约50%的患者存在身材矮小或者癫痫发作(图2)。目前共发现51 种SON变异,主要为移码变异(70.6%)其次为错义变异(13.7%)和无义变异(11.8%),变异均匀的分布于SON基因的外显子上,其中12例患者携带c.5753_5756delTTAG 变异(12/64,18.75%),该位点位于富含精氨酸/丝氨酸功能域,可能为热点变异(图3)。基因型和表型相关性分析未见明显异常,但该结论需要进一步收集患者验证。
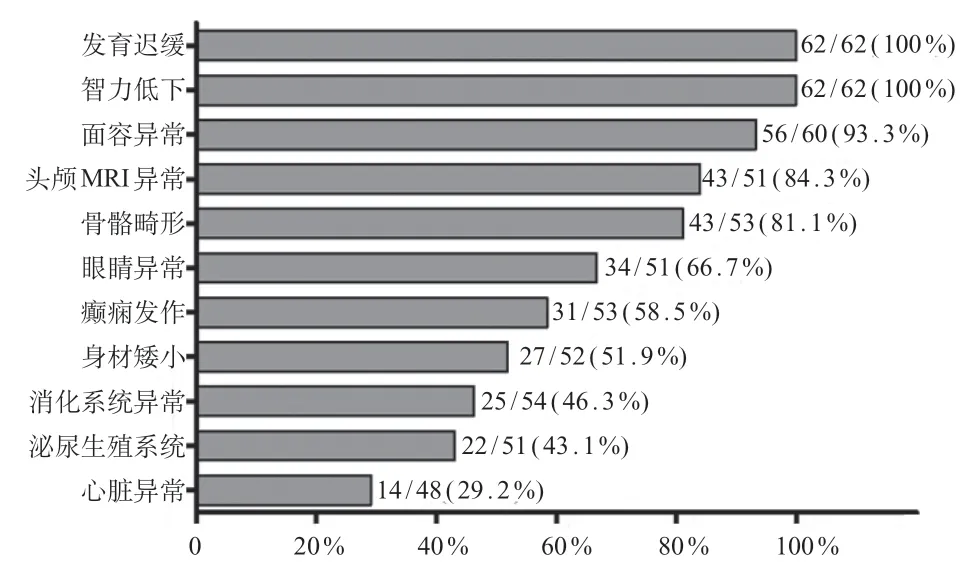
图2 ZTTK 综合征患者的临床表型文献复习
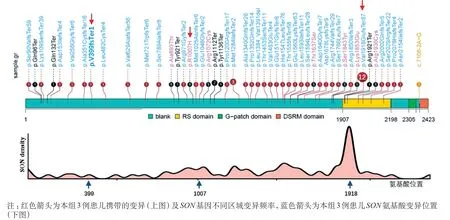
图3 SON 基因与氨基酸变异
SON是RNA加工复合物亚基之一,在mRNA剪接成熟中发挥重要作用[1,7]。Kim等[1]研究发现SON基因敲减的Hela细胞中以及SON基因变异的患者外周血中存在剪接异常的转录本的累积。提取Hela细胞或患者外周血RNA后,检测发现TUBG1、FLNA、PNKP、WDR 62、PSMD 3、PCK 2、PFKL、IDH 2以及TUBA1A等基因均存在异常剪接(内含子保留或外显子跳跃)以及表达水平显著降低。其中TUBG1和TUBA1A基因均与大脑皮质发育不良相关[16-17];文献报道WDR 62与小头畸形相关[18],而约80%的ZTTK患者存在头颅MRI异常。FLNA则关联多种疾病,与脑室旁节结异位、额骨骺发育异常以及FG综合征等[19-20],临床表型异质性很大。推测SON基因异常影响mRNA异常剪接是ZTTK综合征临床累及多系统的原因。
综上所述,ZTTK综合征患者存在发育迟缓、智力低下、面容异常、头颅MRI异常、骨骼畸形及眼睛或视力异常、癫痫发作等多系统异常的临床表现,基因检测有助于早期明确诊断。本研究发现SON基因2个未见报道的新变异,丰富了该基因变异数据库,此外本研究加深临床医师对该疾病的认识。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
上海金属(2021年6期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
中国生殖健康(2020年4期)2021-01-18
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
生物学通报(2019年3期)2019-02-17
中国生殖健康(2018年4期)2018-11-06
百科知识(2015年18期)2015-09-10
湖北农业科学(2014年11期)2014-09-10
