
青岛地区6-丙酮酰四氢生物蝶呤缺乏症患儿基因变异特点及随访分析
2023-02-27 04:28:10钟瑶瑶张立琴陆薇冰刘婷廷
临床儿科杂志 2023年2期
钟瑶瑶 张立琴 杜 玮 陆薇冰 刘婷廷
青岛大学附属妇女儿童医院(山东青岛 266034)
高苯丙氨酸血症(hyperphenylalaninemia,HPA)是由于苯丙氨酸代谢途径中酶缺陷所致的常染色隐性遗传病,根据病因可分为苯丙氨酸羟化酶(phenylalanine hydroxylase,PAH)及其辅酶四氢生物蝶呤(tetrahydrobiopterin,BH 4)缺乏两类[1]。BH 4 不仅是苯丙氨酸羟化酶的辅助因子,也是酪氨酸、色氨酸羟化酶的重要辅助因子,这两种酶参与单胺类神经递质的合成。BH 4 缺乏会导致不同程度的运动障碍、肌张力异常、顽固抽搐、肢体震颤、智能落后等神经症状[2]。参与BH 4 合成或再循环任一酶缺陷均会导致四氢生物蝶呤缺乏症(tetrahydrobiopterin deficieney,BH4D)的发生,其中以6-丙酮酰四氢蝶呤合成酶(6-pyruvoyltetrahydropterin synthase,PTPS)缺乏最为常见[3]。本文对青岛地区26 例PTPS 缺乏症(6-pyruvoyltetrahydropterin synthase deficiency,PTPSD)患儿临床特征进行总结,并研究其基因变异规律及特点,为PTPSD早期诊断、治疗及遗传咨询和产前诊断提供理论依据。
1 资料与方法
1.1 研究对象
以1996 年11 月至2021 年12 月青岛市经新生儿疾病筛查确诊的251 例HPA 患儿为研究对象,所有患儿进行尿蝶呤谱分析、红细胞二氢蝶呤还原酶(dihydropteridine reductase,DHPR)活性测定、BH4负荷试验或基因分析以确诊BH 4 D。BH 4 D 均按2014年《高苯丙氨酸血症的诊治共识》进行诊断,其诊断符合:①血苯丙氨酸(phenylalanine,Phe)水平>120 μmol/L及血Phe与酪氨酸(tyrosine,Tyr)比值(Phe/Tyr)>2.0,诊断为HPA;②PTPS缺乏时,尿新蝶呤(neopterin,N)明显升高,生物蝶呤(biopterin,B)极低,生物蝶呤百分率[B/(B+N)×100%]<10%,血红细胞DHPR活性正常[4]。
1.2 尿蝶呤谱分析和DHPR测定
收集新鲜尿液加入抗坏血酸,浸透专用滤纸上避光晾干,同时收集患儿静脉血制成干血斑标本,所有标本外送上海交通大学附属新华医院新生儿筛查中心进行尿蝶呤谱分析及DHPR活性测定。
1.3 基因分析
用 EDTA 抗凝管抽取患儿外周静脉全血 3 mL,过柱法提取DNA,应用第二代高通量测序技术对BH4D的相关基因进行直接测序。
1.4 随访及智力评估
BH 4 D 患儿定期监测血Phe 浓度,每3~6 个月门诊随访1 次,检测血常规、肝功能,并进行生长发育评估和智能发育检测。随访时间截止至2022 年6月3日。6岁以内患儿采用Gesell儿童发育量表进行智力检测(developmental quotient,DQ),DQ评分低于84 分为智力落后;6 岁以上患儿采用韦氏儿童智力发育量表对智商(intelligence quotient,IQ)进行检测,IQ评分70~79为边缘落后状态,低于70分为智力落后。
1.5 统计学分析
采用SPSS 26.0 统计软件进行数据处理,符合正态分布的计量资料采用x±s表示,非正态分布数据采用中位数(M)表示。
2 结果
2.1 PTPSD发病率及尿蝶呤谱、血DHRP活性测定
青岛市自1996年11月开展新生儿疾病筛查,至2021年12月共筛查1 889 081名新生儿,确诊HPA患儿251例。经尿蝶呤谱、血DHPR活性、BH4D负荷试验或基因检测,诊断BH4D 26例。26例患儿尿蝶呤谱均表现为N/B极度增高[(56.36±44.22)%],B%显著降低[(2.96±2.21)%];血DHPR均在正常范围,故均符合PTPSD诊断。
26例患儿来自23个家庭,其中2对为单卵双胞胎,1对为非同胎生姐妹,PTPSD发病率为12.7/100万(双胎按1例计算)。26例PTPSD患儿中,男14例、女12例,均为足月出生,出生体重为(3.15±0.40)kg,新生儿筛查血Phe平均浓度为(8.76±5.19)ng/dL。
8 例患儿进行B H 4 负荷试验,均在服用BH 4 后出现特征性的快速下降,2 小时内下降(67.63±15.47)%,4小时后下降(86.14±5.43)%至正常水平。
2.2 基因检测结果
19例PTPSD患儿(来自17个家庭)进行基因检测,其中包括单卵双胞胎1对、非同胎生姐妹1对(等位基因相同,均按1例计算)。17个家系的34个PTS等位基因均检出变异,共检出10种变异类型(表1),包含错义变异、剪接变异两种变异类型,其中错义变异发生率为91.17%(31/34),为最常见的变异类型。10 种变异类型中,9 种来源于外显子,1 种来源于内含子。变异位点主要集中在5号外显子,占所有变异类型的67.6%(23/34),本研究中10种变异类型在BIODEF数据中均被报道过。

表1 青岛地区PTPSD患儿PTS基因变异特点
2.3 基因型与表型的关系
19 例进行基因检测的PTPSD 患儿中,7 例确诊年龄>3 月龄,出现不同程度的神经系统症状,临床诊断为严重型PTPSD;3 例确诊后未进行规范治疗,无神经系统症状,诊断为轻型PTPSD。见表2(病例1~10)。
2.4 治疗及随访
19例行基因检测的PTPSD患儿,末次随访中位年龄6.1(1.08~24)岁。所有患儿身高、体重均位于正常同年龄、同性别儿童的P10~P97,提示生长发育正常。16 例接受规范治疗,其中15 例智力发育正常,9 例DQ(92.2±2.5)分,2 例IQ分别是93和94分,4例通过医师综合评价;1例智力落后,因确诊年龄晚所致(例7)。3例未进行规范治疗的患儿(例8~例10),末次智力评估均正常。见表2。
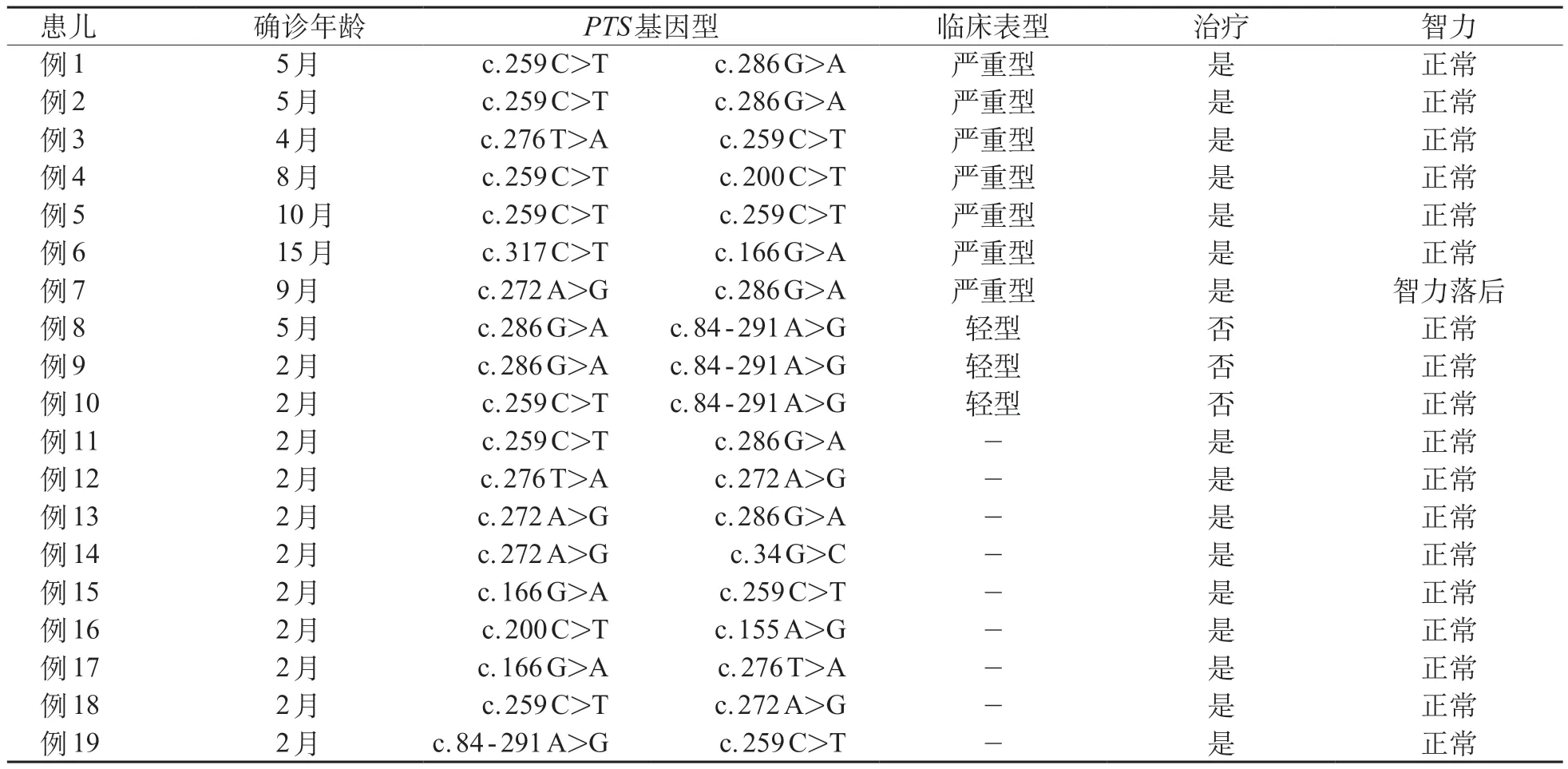
表2 19例PTPSD患儿临床特征和基因型
7 例未行基因检测的患儿,1 例1 岁时因肺炎死亡,2 例失访,2 例智力检测正常,2 例智力落后(1 例生后3 月龄确诊PTPSD,因家庭原因未规范服用治疗药物,1 例确诊年龄2 岁,就诊时即存在智力落后)。
3 讨论
BH 4 D 是一类具有高度遗传异质性的疾病,约占HPA人群的1%~20%[2]。我国自1981年开展新生儿HPA 筛查,后逐渐引进尿蝶呤分析、BH 4 口服负荷试验、DHPR 活性检测、基因分析等,以鉴别各型BH4D。青岛市新生儿疾病筛查中心自1996年开展新生儿筛查起即注意BH4D的鉴别诊断,2005年首次对本中心诊断的7例BH4D患儿临床特征进行总结报道[5],为BH4D的诊治提供了临床经验。
BH4D在不同地区、不同种族HPA中发生率具有差异,中东、亚洲人群发生率较高[6-7],在我国也有显著的地区差异。我国一项来自上海、徐州、广西和南京等地区筛查研究显示,800多例HPA患者中,BH4D占8.2%[8],而河南地区发生率仅为4.1%[9]。本研究中BH4D占HPA患儿的10.3%,高于我国其他地区报道。BH4D全球发病率尚不明确,不同国家之间存在很大差异。我国一项全国范围的研究发现BH4D发病率为3.8/100万,其中东部、北部地区发病率较高(5.9/100万、4.1/100万),南部和西北部地区发病率分别为1.6/100万、1.7/100万[10]。本研究青岛地区BH4D(均为PTPSD)发病率为12.7/100万,明显高于既往文献报道。
目前PTPSD是最为常见BH4D类型。国外研究显示PTPSD占BH4D的57%[2],而中国大陆人群中该比例可高达96%[11-12]。本研究中BH4D患儿均为PTPSD,与文献报道基本一致。PTS基因位于第11号染色体(11p2.2.3),共包含6个外显子,人PTS基因cDNA 在1992 年首次被克隆[13]。截止2022 年6 月,BIOPKU 数据库已收录198 种PTS基因变异,其中c.155A>G、c.259C>T、c.272A>G和c.286G>A是中国人群最常见的变异。本研究中,青岛市PTPSD患儿PTS基因变异以复合杂合变异为主,具有明显的热点变异,c.259 C>T 是主要的变异类型,其次为c.286G>A,发生率为18%,这与我国河南、四川等地区变异特征相一致[9,12]。同时,c.272 A>G 也是青岛地区BH4D人群的热点变异之一,变异发生率为13%,河南、四川、南方等地区的报道[9,11-12]。BIOPKU数据库显示PTS变异位点主要位于5号和6号外显子,本研究中PTS基因变异频率最高的也是5号外显子,对PTS基因外显子5进行基因变异位点检测,可大大提高基因诊断的效率,降低成本,可早期诊断PTPSD患儿。
BH4D临床表现的严重程度受多种酶活性影响,有多项研究对PTS基因不同变异类型进行功能验证,已证实c.200C>T、c.259C>T、c.286G>A导致PTPS酶活性为野生型的63%、52%和10%,多导致严重型PTPSD[14-15]。本研究中,6 例严重型PTPSD 患儿均携带该3种变异,与文献报道一致。由于本研究中心尚未开展脑脊液神经代谢产物的测定,且新生儿筛查早期患儿多无临床症状,故BH4D轻型与严重型分型困难。本研究中8 例进行基因检测的PTPSD尚未明确临床分型,无法明确其基因型和表型之间的相关性,但根据已统计的基因型与表型的关系及临床随访结果显示,携带c.166 G>A、c.272 A>G、c.276T>A基因型可能与轻型PTPSD有关[16-17],而携带有c.155A>G、c.317C>T、c.200C>T、c.259C>T、c.286G>A的变异基因型则与严重型PTPSD有关[14-16]。
值得注意的是,本研究中3 例携带c.84-291 A>G 变异的患儿未接受规律药物治疗,目前随访血苯丙氨酸浓度及智力发育均正常。c.84-291 A>G位于PTS基因的内含子1,影响mRNA的剪接过程,该变异可产生3种剪接mRNA:正常长度、外显子3缺失、79 bp假外显子插入,其中正常长度的mRNA占23.8%~37%[18-19]。由于该变异仍能产生正常长度的拼接产物,能够维持部分PTPS的酶活性,从而导致轻型PTPSD。我国学者报道3例携带c.84-291 A>G变异的PTPSD患儿在诊断早期即停止药物治疗,随访过程中血苯丙氨酸维持正常,与本研究结果相一致[19]。
BH 4 D 患儿在新生儿期多无任何临床表现,与苯丙氨酸羟化酶缺乏症难以区分,当给予低/无Phe奶粉喂养后血Phe 水平可很快下降,误以为治疗有效,而一旦出现神经系统损害,易导致严重的智力障碍等后遗症[20]。目前随着BH 4 D 诊疗水平的提高,多数患儿在3 个月内即可进行诊断。本研究中近10年出生的BH4D患儿有92%(12/13)在生后2个月确诊,早期给予相应治疗,目前该部分患儿智能测试结果均在正常范围内,提示早期诊断及治疗可明显改善患儿预后。目前多采用补充BH4及神经递质前体左旋多巴、5-羟色氨酸联合治疗。但在开展新生儿筛查的初期诊断,部分患儿因诊断当时无BH4药物等联合治疗或因经济原因而放弃。青岛市自1996年11月开展新生儿疾病筛查工作至今已经历26年,BH4D从诊断难到治疗难,从确诊患儿的缺药、少药、求药到2012年在全国率先将治疗药物纳入医疗保险体系,解决了家庭沉重的经济负担,使患儿得以长期终生治疗。青岛地区诊断的第1 例病例目前年龄24岁,已经步入社会,正常工作生活。患儿的健康成长得益于新生儿疾病筛查-诊断-治疗及其长期随访-救助的一体化管理体系。
综上所述,1996-2021年25年间青岛市新生儿BH4D平均发病率为12.7/100万,最常见的变异基因为PTS,基因检测有助于BH4D的诊断,对已经明确基因变异的患儿进行家系分析和遗传咨询,有利于提高出生人口素质。早期诊断,尽早使用BH 4 和神经递质前体进行治疗,可以有效降低BH4D患儿神经系统损害,减少后遗症的出现,达到降低残疾儿发生率的目的。通过新生儿疾病筛查,早期诊断和治疗是患儿健康成长的关键。建立完善的新生儿疾病筛查-诊断-治疗-长期随访-救助的一体化管理体系是作好出生缺陷第三级预防,降低残疾儿发生,提高人口素质的有力措施。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
世界最新医学信息文摘(2021年24期)2021-04-29 03:47:14
中国生殖健康(2020年4期)2021-01-18 02:58:10
中国生殖健康(2018年4期)2018-11-06 07:12:16
小天使·二年级语数英综合(2017年4期)2017-04-18 17:29:21
小天使·四年级语数英综合(2017年4期)2017-04-18 09:15:43
分析测试学报(2015年4期)2016-01-13 06:18:25
河北科技大学学报(2015年6期)2015-03-11 16:16:54
少年文艺·开心阅读作文(2014年5期)2014-10-08 16:11:31
湖北农业科学(2014年11期)2014-09-10 18:06:07
