
金属有机框架化合物衍生Mn2O3催化过一硫酸盐降解左氧氟沙星*
2023-02-22 05:44韩晓琳修光利
环境污染与防治 2023年2期
李 帅 韩晓琳 张 巍# 修光利
(1.国家环境保护化工过程环境风险评价与控制重点实验室,上海 200237;2.华东理工大学资源与环境工程学院,上海 200237;3.上海市环境保护化学污染物环境标准与风险管理重点实验室,上海 200237;4.上海污染控制与生态安全研究院,上海 200092)
左氧氟沙星(Levo)因具有广谱抗菌性而被广泛应用于医药行业[1]。Levo在生物体内不易降解,因此大量的Levo会以母体形式排放到环境中[2],易诱导产生耐药细菌,然而传统的生化处理难以将其进行有效降解。过一硫酸盐(PMS)高级氧化技术能有效降解环境中的有机污染物[3],但需要合适的方法催化PMS才能高效降解有机污染物。PMS的催化方法有加热、超声、光、过渡金属离子、金属氧化物等[4-7],其中金属氧化物催化相较于其他方法具有催化剂可重复利用、操作简便、反应稳定等优点,极具发展潜力[8]。锰氧化物被认为是一种优良的金属氧化物催化剂[9],不仅因为锰储量丰富、价格低廉,而且相对其他金属而言更加环境友好。
在各种锰氧化物中,Mn2O3催化PMS的效率高且所需活化能低[10]。近年,以金属有机框架化合物(MOFs)衍生制备锰氧化物得到越来越多的研究人员关注[11-12]。MOFs原位高温热解制备的Mn2O3具有不易团聚的优点,有利于锰氧化物表面的活性位点与PMS和有机污染物充分接触[13]。一般认为,PMS非均相催化体系的催化机理为自由基路径,但也有研究者认为单线态氧(1O2)、电子介导等非自由基路径也存在于PMS非均相催化体系中[14-15]。非自由基路径具有选择性高、氧化剂利用率高、pH适用范围广、不产生有毒卤素副产物等优点[16]。目前,Mn2O3催化PMS降解有机污染物的非自由基路径报道甚少。
本研究用MOFs衍生制备Mn2O3作为催化剂,用于催化PMS降解Levo,对催化剂的形貌结构进行了表征,探讨了反应过程中初始pH、水体中无机阴离子和腐殖酸(HA)对Mn2O3催化PMS降解Levo的影响,并通过淬灭实验、电子顺磁共振(EPR)实验和计时电流检测判断PMS非均相催化体系的反应机理,为基于非自由基路径的新型PMS催化体系构建和相关催化剂研发提供新的思路和参考。
1 材料与方法
1.1 试 剂
四水合氯化锰(纯度>99.99%);Levo(纯度>98%);过一硫酸氢钾(纯度>98.5%);苯醌(p-BQ,纯度>98%);氢氧化钠(纯度>96%);浓盐酸(质量分数36%~38%);叔丁醇(TBA,纯度>98%);均苯三甲酸(纯度>99%);甲醇(纯度>99.5%);乙醇(EtOH,纯度>99.5%);氯化钠(纯度>99.5%);磷酸二氢钠(纯度>98%);HA(纯度>90%);糠醇(FFA,纯度>99%);2,2’-联氮双(3-乙基苯并噻唑啉-6-磺酸)二铵盐(ABTS,纯度>98%);醋酸(纯度>95%);醋酸钠(纯度>99%);5,5-二甲基-1-吡咯啉-N-氧化物(DMPO,纯度>97%);2,2,6,6-四甲基哌啶(TEMP,纯度>95%);磷酸(纯度>99.99%);去离子水。
1.2 仪 器
Tristar Ⅱ 3020 型比表面积孔隙度分析仪(BET)、2695型高效液相色谱(HPLC)仪、D/max-RB型X射线衍射(XRD)仪、S-3400N型扫描电子显微镜(SEM)、PHI5300型X射线光电子能谱(XPS)仪、CHI 760E型电化学工作站、SHO5-3G型恒温磁力搅拌器、DZF-6021型真空干燥箱和SX2-4-10型马弗炉。
1.3 催化剂制备
首先,借鉴改进的共沉淀方法[17]把2.26 g氯化亚铁替换为2.26 g四水合氯化锰合成锰基MOFs(Mn-MOFs),用去离子水和EtOH洗涤3次后,置于真空干燥箱中于60 ℃下干燥12 h。然后,采用高温煅烧方法[18]在空气气氛下将Mn-MOFs置于马弗炉中于700 ℃下热解制得Mn2O3,用去离子水和EtOH洗涤3次后,置于真空干燥箱中于60 ℃下干燥12 h,备用。
1.4 实验方法
移取100 mL 1 mg/L的Levo溶液于250 mL锥形瓶中,调节初始pH至预定值,加入适量Mn2O3和PMS使投加量分别为0.04 g/L、0.15 mmol/L,按照一定的时间间隔取1 mL反应液并与1 mL甲醇混合后用0.45 μm滤膜过滤,使用HPLC仪对Levo浓度进行测定。
1.5 分析方法
XRD测试以Cu-Kα为衍射源,管电压为40 kV,电流为40 mA,波长为1.540 6 Å,扫描速率为4°/min,扫描范围为5°~75°。
XPS测试以Al-Kα(1 486.6 eV)为激发源,束斑大小为400 μm,分析室真空度小于5.0×10-5Pa,测试电压为12 kV,电流为6 mA。
SEM的工作电压为15 kV,放大倍数为15 000倍。
将材料置于150 ℃ N2氛围下脱气处理4 h后在-196.15 ℃条件下进行BET N2物理吸附/脱附测定。
PMS浓度使用ABTS作为显色剂通过分光光度法[19]测定。
Levo浓度测定的HPLC条件:流动相为甲醇和0.1%(质量分数)磷酸缓冲液(按磷酸和水体积比35∶65配制),检测波长为280 nm。Levo降解采用伪一级动力学方程(见式(1))进行拟合。
(1)
式中:ct、c0分别为t时刻和初始时刻Levo的质量浓度,mg/L;k为伪一级动力学常数,min-1。
2 结果与讨论
2.1 MOFs衍生Mn2O3表征结果
所制备的催化剂样品的XRD图谱见图1,在2θ为23.13°、32.95°、38.23°、45.17°、49.34°、55.18°、65.80°处的衍射峰与Mn2O3(JCPDS No.41-1442)的(211)、(222)、(400)、(332)、(431)、(440)、(622)晶面吻合,表明所制备催化剂为Mn2O3。
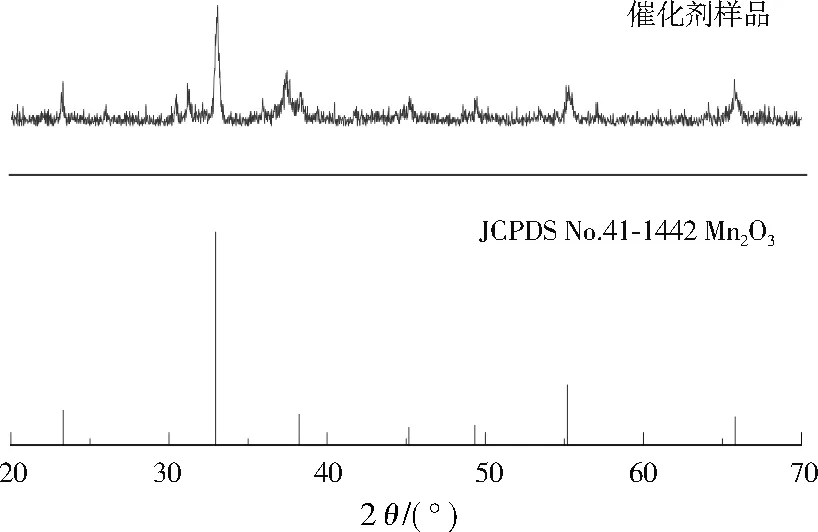
图1 所制备催化剂的XRD图谱Fig.1 XRD pattern of the prepared catalyst
使用XPS仪分析催化剂样品表面的原子组成和化学状态,发现存在明显的Mn 2p(641.4 eV)和O 1s(529.3 eV)元素峰(见图2),质量分数分别为65.9%、34.1%,与Mn2O3的质量分数一致,进一步表明Mn2O3被成功合成。
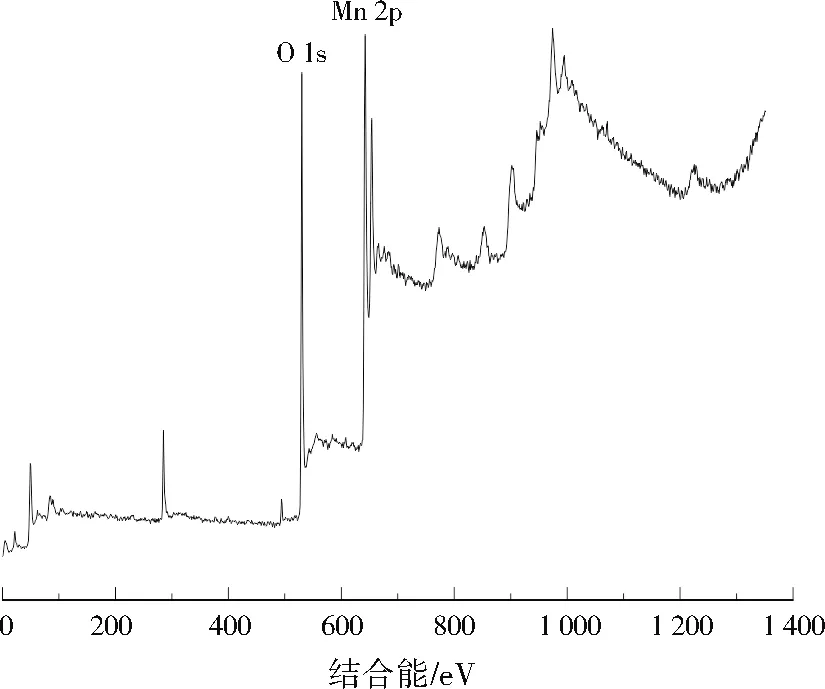
图2 所制备催化剂XPS全谱Fig.2 XPS full spectrum of the prepared catalyst
图3的SEM照片显示了Mn2O3颗粒的微观形貌,为棒状结构,表面光滑,晶体长度为300~3 000 nm,宽度为6~600 nm,属纳米级材料。

图3 Mn2O3的SEM照片Fig.3 SEM image of Mn2O3
同时BET分析得到,Mn2O3的比表面积为2.9 m2/g,与文献[19]中用类似制备方法得到的Mn2O3材料相一致。
2.2 不同反应体系对Levo降解效果的影响
在初始pH为6.7的条件下,比较了Mn2O3催化PMS、单独PMS和单独Mn2O33个反应体系降解Levo的效果差异。由图4可知,单独PMS和单独Mn2O3体系均不能有效降解Levo,45 min时去除率分别只有11.8%、19.1%,说明单独PMS的直接氧化效率较低,单独Mn2O3的吸附/氧化作用对去除水中Levo的效果也不明显。而当Mn2O3和PMS同时存在时,45 min时Levo的去除率可以达到71.2%,这说明Mn2O3可以高效催化PMS,促进Levo降解。Levo在Mn2O3催化PMS、单独PMS和单独Mn2O3反应体系内的伪一级动力学常数分别为0.033 7、0.003 8、0.006 3 min-1,同文献中锰矿渣-N3H(0.029 min-1)[20]、550-MnO@MnOx(0.032 1 min-1)[21]和CuFe2O4(0.028 7 min-1)[22]等催化剂催化PMS降解Levo的反应速率常数相比,本研究制备的Mn2O3能在催化剂和PMS投加量均较低的条件下高效降解Levo。
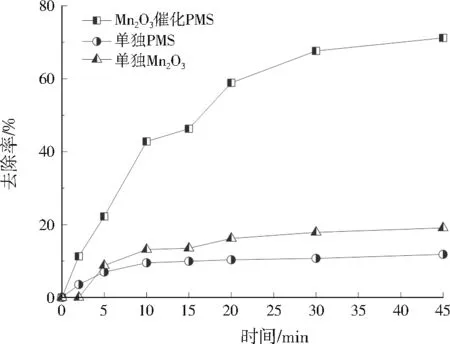
图4 不同反应体系降解Levo的性能Fig.4 Performance of different systems to degrade Levo
2.3 初始pH、无机阴离子和HA对降解效果的影响

图5 初始pH、无机阴离子和HA对Mn2O3催化PMS降解Levo效果的影响Fig.5 The effect of initial pH,inorganic anions and humic acid on the removal of Levo degradation by PMS catalyzed by Mn2O3
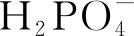
2.4 降解机理研究
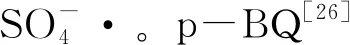
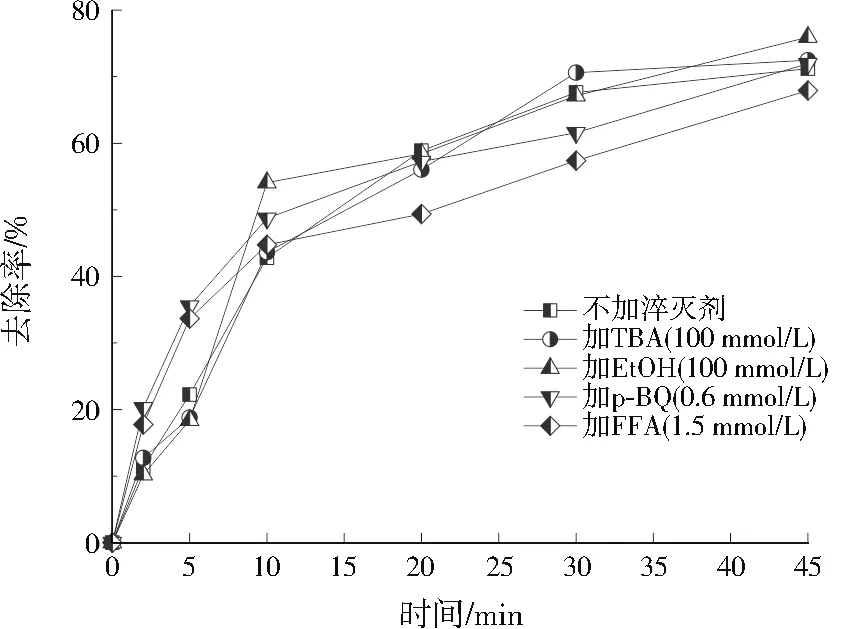
图6 淬灭实验结果Fig.6 Results of quenching experiment
使用XPS仪分析反应前后Mn2O3催化剂表面的化学状态变化。Mn 2p的XPS精细图谱如图7(a)所示,反应前后Mn 2p1/2和Mn 2p3/2峰的结合能均分别为641.6、653.0 eV,Mn 2p1/2和Mn 2p3/2间的分裂能为11.4 eV,与Mn2O3的分裂能一致[28]。O 1s的XPS精细图谱如图7(b)所示,529.3、530.9 eV处的谱峰分别对应Mn—O—Mn和材料外表面的O—H[29]。催化剂反应后,Mn—O—Mn键强度有所减弱,摩尔分数由反应前的64.5%略微减少为反应后的62.1%,可能是因为Mn2O3的少量晶格氧参与了1O2的生成,从而导致Mn—O—Mn强度减弱。一般来说,金属催化剂通过金属价态的相互转化产生电子,攻击PMS的O—O键,从而促进自由基产生。在此过程中可以发现金属催化剂上金属价态的变化[30-32]。本研究中,反应前后锰的价态均为Mn(Ⅲ),未发生改变,这说明PMS被催化并不是因为锰价态改变后释放电子导致的,侧面支持了该反应体系中电子介导机理的可能性。

图7 反应前后Mn2O3催化剂的XPS精细图谱Fig.7 XPS fine spectra of Mn2O3 catalysts before and after reaction
若Mn2O3催化PMS反应体系中确实存在电子介导机理,则Mn2O3与PMS将首先形成复合物,然后促使吸附在Mn2O3表面的Levo分子上的电子通过Mn2O3向PMS转移[25]8-9。因此研究比较了Mn2O3催化PMS、PMS降解Levo和Mn2O3催化PMS降解Levo 3种情况下的PMS降解率,图8(a)的结果显示,3种情况下45 min时PMS的降解率依次为19.4%、10.2%、28.8%,这就预示着Mn2O3催化PMS降解Levo的过程中可能存在电子介导机理。同时计时电流测得加入PMS和Levo后的电流变化,图8(b)的结果显示,当PMS加入(200 s)后,电流由4.26 μA降至1.27 μA,当Levo加入(300 s)后,电流突增至4.78 μA,这说明PMS和Levo的加入对电流输出有着显著的影响,与文献[26]报道的现象一致,表明PMS、Mn2O3表面和Levo之间存在电子转移。

图8 电子介导机理验证Fig.8 The vallidation of electron transfer mechanism
综上所述,可以判断Mn2O3催化PMS降解Levo过程中电子介导机理起着重要作用,同时还存在1O2的贡献。与单一电子氧化过程不同,在电子介导的氧化反应过程中,PMS与Mn2O3形成的复合物(电子受体)从Levo(电子供体)中提取两个电子,从而实现Levo的快速降解[33]。Mn2O3催化PMS降解Levo过程的机理可总结为图9。
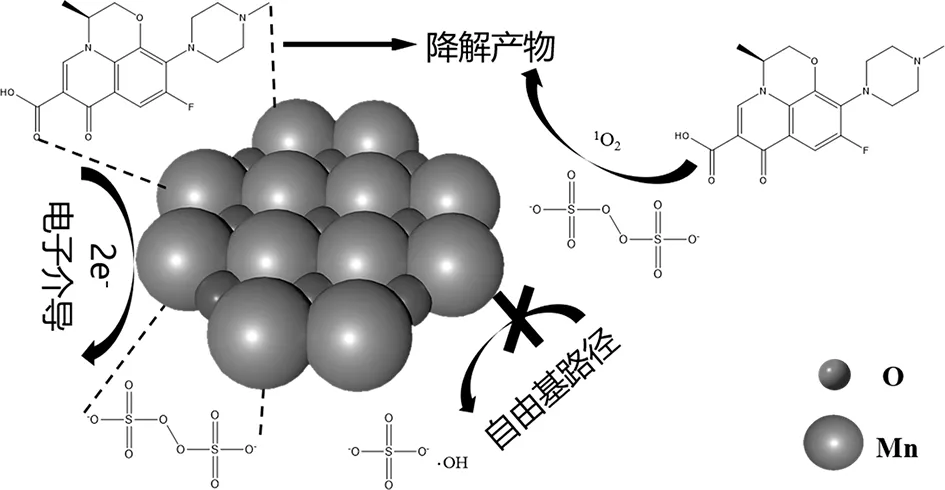
图9 Mn2O3催化PMS降解Levo的反应机理Fig.9 The reaction mechanism of Levo degradation by PMS catalyzed by Mn2O3
3 结 论
采用高温煅烧Mn-MOFs的方法成功制备了纳米Mn2O3,其在PMS存在的条件下体现出了对Levo良好的降解性能,降解过程符合伪一级动力学,相应伪一级动力学常数为0.033 7 min-1。Mn2O3催化PMS反应体系能够在宽的初始pH(3.0~9.0)范围内高效降解Levo,无机阴离子和HC对反应体系的影响不大,Cl-甚至能提升Mn2O3催化PMS反应体系对Levo的降解效率。自由基淬灭实验、EPR实验和计时电流检测证明,Mn2O3催化PMS降解Levo过程中电子介导机理起着重要作用,同时还存在1O2的贡献,均属于非自由基路径。
猜你喜欢
建材发展导向(2021年14期)2021-08-23
粉末冶金技术(2021年3期)2021-07-28
陶瓷学报(2020年6期)2021-01-26
中国煤层气(2019年2期)2019-08-27
中学生数理化·中考版(2018年11期)2019-01-31
教学考试(高考化学)(2018年5期)2018-12-06
童话世界(2017年29期)2017-12-16
环境与可持续发展(2017年2期)2017-04-06
中学生数理化·高二版(2016年6期)2016-05-14
应用化工(2014年11期)2014-08-16
