
阿尔茨海默病中蛋白质相互作用对β-分泌酶的调节机制
2023-02-21 10:00刘聪聪王培昌王亚琦
中国生物化学与分子生物学报 2023年1期
刘聪聪, 王培昌, 王亚琦
(首都医科大学宣武医院检验科, 北京 100053)
阿尔茨海默病(Alzheimer’s disease, AD)是一种与年龄及特定神经病理学相关的慢性退行性疾病。AD的典型病理特征包括淀粉样斑块沉积、神经纤维缠结的形成以及神经元的凋亡。疾病的进程呈进行性,伴随着神经元的死亡,患者逐渐出现认知功能障碍、身体各项机能丧失、失去自理能力,最终导致死亡[1]。截至2019年底,我国60岁及以上老年人约有2.54亿,老年人患痴呆人数超过1千万,老年期痴呆患病率约3.99%[2],在各种病因所导致的痴呆病例中,诊断为AD的病例超过600万,占比高达50%~75%[3]。然而,目前尚无有效的治疗方案,其原因在于AD的发病机制不明晰。因此,探究AD发病机制以及治疗方法成为老年医学的重要课题,β-分泌酶(β-site APP cleaving enzyme 1,BACE1)作为AD发病环节的重要一环,值得进行深入探讨。蛋白质之间的相互作用是蛋白质发挥其生物学功能的重要途径,参与细胞信号转导、细胞增殖凋亡等调控生命活动的重要过程,探究BACE1及其与相互作用蛋白质之间的相互作用,能够为AD的发病分子机制以及药物研发带来启示。
1 β-分泌酶的生物学功能
在人体内,β-淀粉样蛋白前体蛋白(amyloid precursor protein, APP)存在两条降解途径:非淀粉样降解途径和淀粉样降解途径,正如Fig.1所示。正常生理条件下,APP以非淀粉样降解途径为主,而在AD中,APP通过淀粉样降解途径生成β-淀粉样蛋白(β-amyloid protein, Aβ)增多。β-分泌酶(β-site APP cleaving enzyme 1, BACE1)是胃蛋白酶家族的I型天冬氨酸蛋白酶,是催化APP分解成为Aβ关键步骤的限速酶[4],BACE1能特异性识别并切割APP 671和672之间的位点,裂解APP产生可溶性的sAPPβ和含有99个氨基酸残基的跨膜片段C99或CTF-β,γ-分泌酶进一步切割C99产生了Aβ40与Aβ42两种不同长度的产物[5]。Aβ42更易聚集形成寡聚体,引起AD的特征性病理改变,形成脑内淀粉样斑块以及tau蛋白磷酸化后神经原纤维缠结[6]。

Fig.1 Proteolytic processing and cleavage sites of APP (A) Non-amyloidogenic pathway. APP is cleaved into C83 and sAPPα by α-secretase, and C83 is further cleaved into P3 and AICD by γ-secretase, which is the main pathway of APP degradation. (B) Amyloidogenic pathways. APP is cleaved by β-secretase into C99 and sAPPβ. Due to the different cleavage sites of γ-secretase, C99 is further cleaved into Aβ40 and Aβ42
在AD患者的大脑和体液中,检测到BACE1的浓度和活性增加。AD患者的尸检结果显示,其脑组织中BACE1水平增高[7]。该结果验证了BACE1在AD病理生理学中发挥关键作用的淀粉样级联假说。BACE1催化的淀粉样斑块的形成是AD早期发病的机制。BACE1水平的升高发生于神经元凋亡之前,并正反馈地推动AD病程的进展,是AD发生的早期信号[8],抑制BACE1的表达量及其活性,能减少致病的淀粉样斑块的聚集,从而进一步延缓AD的病理进程。BACE1作为淀粉样斑块形成起始环节的重要一环,必然会成为早期AD的治疗靶点[9]。
2 β-分泌酶的合成与修饰
完整的BACE1包含5个功能结构域。正如Fig.2所示,分别是信号肽结构域、前结构域、催化结构域、胞质结构域和跨膜结构域。每个功能结构域都在BACE1的合成修饰、活性表达中发挥重要的作用。BACE1基因位于11号染色体,包含9个外显子和8个内含子[10],经转录和翻译形成完整肽链。BACE1首先在内质网上合成含有501个氨基酸的酶原,经过乙酰化和糖基化修饰,折叠形成稳定的空间构象。此时,催化结构域经过折叠,使BACE1形成有活性的蛋白酶,进而向高尔基体运输。在高尔基体中,用于内质网定位的前结构域及信号结构域,被前蛋白转化酶(Furin)切除,并形成成熟的BACE1。成熟的BACE1经运输到达细胞膜,并内化进入内体。其中,跨膜结构域的棕榈酰化与胞内结构域的磷酸化修饰,对于BACE1锚定脂阀、调节BACE1质膜到内体的循环发挥了重要作用[11]。经过上述一系列糖基化、磷酸化、棕榈酰化和乙酰化修饰[4],BACE1广泛表达在神经系统的各类细胞中。其中,神经元、少突胶质细胞和星形胶质细胞表达最为丰富[12]。

Fig.2 The schematic diagram of human BACE1 structure The BACE1 protein contains five domains, namely signal peptide domain, pro domain, catalytic domain, transmembrane domain and cytoplasmic domain. The pro domain and signal domain are excised by Furin to form mature BACE1
当前的医疗水平无法治愈AD,早预防、早发现与早治疗对缓解病情的进展及严重程度至关重要[13]。目前,用于AD监测的检验方法有脑组织活检、血清与脑脊液(cerebrospinal fluid, CSF)Aβ1-42、T-tau、P-tau的检测等[14]。但在临床实践中,常规开展的标志物仅有CSF Aβ42、T-tau和P-tau[15]。最新研究显示,血浆、血清BACE1活性检测诊断AD准确率达到了77%,敏感性/特异性:73/70%,可作为候选生物标志物辅助AD的早期诊断[16]。早年的研究主要关注于BACE1抑制剂的研发,但因为BACE1底物众多、治疗无效或者是抑制剂有较强的毒副作用停止了临床试验[17]。目前,BACE1抑制剂的研究陷入瓶颈,以BACE1为治疗靶点的研究需要找到一个新的方向。
3 在AD疾病进程中β-分泌酶相互作用蛋白质对其的调节作用
除了目前已知的miR-107、miR-29c、miR-339-5p、miR-186、miR-195、miR-135b、miR-135a、miR-124、miR-298、miR-328、miR-361-3p等非编码mRNA能对BACE1的表达进行负调控外[18-23],正如Fig.3所示,BACE1相互作用蛋白质可以在BACE1转录水平对BACE1启动子活性、mRNA水平进行调控。同时,也会在BACE1转录后翻译修饰环节对BACE1在细胞内分布和转运进行调节,从而影响BACE1的酶活性。相互作用蛋白质也可以通过改变BACE1构象以及与底物竞争性结合的方式调节BACE1的活性。因此,探讨BACE1相互作用蛋白质对BACE1调节的具体机制,对深入研究BACE1的表达与功能具有重要意义。
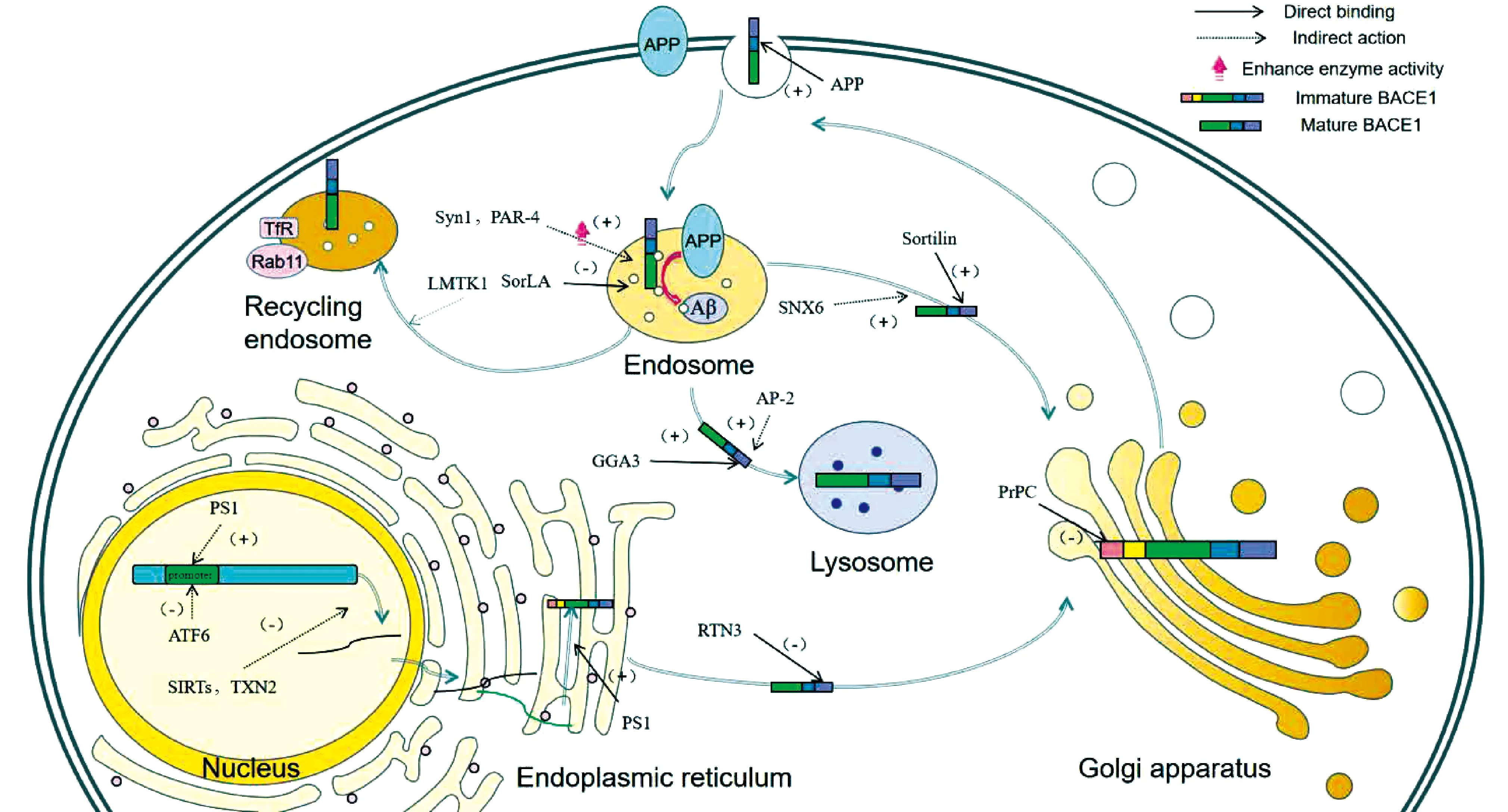
Fig.3 Action sites of BACE1 interacting proteins ATF6, Sirtuin 1 and TNX2 regulate the transcription level of BACE1. APP, Sortilin, PrPc, LMKTKI and SNX6 can modulate the distribution and intracellular transport of BACE1. Syn1 and PAR-4 regulate activity of BACE1. Both Ap-2 and GGA3 regulate the degradation of BACE1. RTN3 and SorLA can regulate the cleavage of APP by BACE1
3.1 BACE1相互作用蛋白质通过调节BACE1的转录过程影响其生成
研究表明,BACE1主要表达于神经系统,在神经元中BACE1启动子活性要高于非神经细胞,转录激活因子6(activating transcription factor 6, ATF6)是一种位于内质网膜上的Ⅱ型跨膜蛋白质[24],过表达的AFT6能够通过与其他影响因子相互作用抑制BACE1基因启动子活性,从而抑制BACE1的表达,同时又能促进α-分泌酶的表达[25]。沉默信息调节因子(sirtuins, SIRTs)是重要的抗衰老因子,与神经系统疾病有关,沉默信息调节因子1(sirtuin 1, SIRT1)通过过氧化物酶体增殖物激活受体γ(peroxisome proliferator-activated receptor gamma, PPARγ)和过氧化物酶增殖物激活受体γ辅助激活因子-1α (Peroxisome proliferator-activated receptor-γ co-activator-1α PGC-1α)去乙酰化,降低BACE1的转录,从而减少BACE1的生成[26]。沉默信息调节因子2(sirtuin 2, SIRT2)是其主要的亚型,在脑细胞中高度表达[27]。有研究显示,SIRT2/内质网蛋白4B(reticulon4B, RTN4B)/BACE1病理途径是AD发生的重要因素。SIRT2的降低可以提高RTN4B水平,从而降低BACE1水平。其机制是SIRT2通过催化RTN4B去乙酰化,驱动RTN4B的降解。RTN4B与内质网蛋白3(reticulon3, RTN3)同属于RTN家族,具有相同的C-端结构域,RTN4B与RTN3通过相同的负调控方式与BACE1相互作用并影响其表达,减少BACE1的产生[28]。硫氧还蛋白-2(thioredoxin-2, TXN2)是一种线粒体蛋白质,其表达量随着年龄的增长而降低。TXN2沉默或过度表达能选择性地增加或减少BACE1的转录,但不改变参与APP催化处理的其他酶的水平[29]。早老蛋白-1(presenilin-1, PS1)与BACE1结合后,能够增强BACE1启动子活性,增加mRNA的转录以及BACE1的表达水平,正向调节BACE1的合成、修饰与表达[30]。研究显示,DNA甲基化能够调节BACE1的表达,其原因可能是由于PS1转录因子结合到BACE1启动子区域CpG位点,从而增加导致BACE1转录水平增高[31]。在重度AD患者的皮质区神经元中发现了大量CpH位点的低甲基化增强子,这可能会上调BACE1转录水平[32]。
3.2 BACE1相互作用蛋白质通过调节BACE1的细胞内转运影响其与APP共定位
细胞是一个高度有序的结构,蛋白质只有在特定的区域中才能有效发挥其功能活性。BACE1的神经元转运和细胞定位对于BACE1活性和突触中Aβ的产生至关重要。研究发现,BACE1主要在内体中对APP进行切割,在BACE1质膜-早期内体转运期间与APP存在空间的分隔,仅在BACE1与APP在早期内体有共同定位时,BACE1才能对APP进行切割,而BACE1的不同亚细胞定位在一定程度上决定了BACE1对APP的切割作用。BACE1部分相互作用蛋白质以直接或间接结合的方式参与了BACE1的细胞内运输的调节。APP可以与BACE1跨膜结构域的相互结合,促进BACE1的内吞作用。分拣蛋白Sortilin属于Vps10p受体家族,是一种跨膜蛋白质,Sortilin的胞质结构域尾部与BACE1胞外结构域相互作用,参与调节BACE1的亚细胞分布以及逆向转运[33]。除了上文所提到的PS1能够与BACE1结合增强BACE1启动子活性外,它还能够调节BACE1的胞内运输,促进其向内质网层迁移,促进BACE1更快地发挥其功能活性[30]。磷脂爬行酶1(phospholipid scramblase 1, PLSCR1)能够与BACE1尾部的双亮氨酸残基结合并相互作用,影响BACE1在脂筏上的分布和募集,调节BACE1的细胞内分布[34]。细胞型朊蛋白(cellular prion protein, PrPC)可以与较低分子量的未成熟的BACE1直接结合,结合部位为定位高尔基体的前结构域,这种相互作用增加了BACE1在高尔基体中的含量,抑制其向细胞表面和核内体的运输,抑制BACE1对APP的切割,减少具有神经毒性的Aβ的产生[35],同时PrPC和Aβ的错误折叠会引起神经元树突棘的缺失等病理性改变[36, 37]。
BACE1其他相互作用蛋白质以非结合的方式调节BACE1的细胞内转运。Rab基因编码的Rab蛋白家族在人体内分布广泛。研究证明,Rab基因突变与神经退行性疾病的发生发展有着高度密切的联系。Rab39B蛋白属于Rab GTP酶家族,Rab39B的缺失会使BACE1水平增高,过表达的Rab39B通过特异性改变细胞内BACE1的转运和分布来降低细胞膜上BACE1的水平[38]。Rab11与细胞凋亡相关的酪氨酸激酶(lemur tail kinase 1, LMTK1)共同调节BACE1的转运,BACE1与Rab11阳性内体紧密结合,并且其定位受LMTK1A激酶活性调控。LMTK1是一种在神经元中大量表达的内体丝氨酸/苏氨酸激酶,能够通过调控Rab11依赖的BACE1内体定位[39]。分选连接蛋白6(sorting nexin 6, SNX6)与分选连接蛋白12(sorting nexin 12, SNX12)同属于含有PX域的SNX家族,该家族对细胞内的膜泡运输发挥了重要作用[40],SNX6通过负向调节BACE1在内吞环节的逆行转运以及BACE1的基础表达水平来调节BACE1介导的Aβ的生物学转化[41]。与SNX6相同的是,SNX12过表达引起的Aβ减少也是通过影响BACEl在细胞内的转运,进而影响对APP的剪切[42]。衔接蛋白-2(adaptor protein 2, AP-2)能通过调控BACE1向溶酶体的运输,来调节APP裂解成为Aβ的过程。在特异性敲减AP-2的小鼠中观察到,BACE1在突触中向溶酶体的转运发生阻滞,自噬水平降低,BACE1的活性并未发生改变,而BACE1依赖的APP切割水平升高,从而引起Aβ的堆积和淀粉样斑块的形成[43]。
3.3 BACE1相互作用蛋白质通过调节BACE1的活性影响其对APP的切割作用
BACE1相互作用蛋白质可以对BACE1的活性进行调节。突触蛋白-1(synapsin 1, SYN1)参与BACE1对APPβ位点切割的调节。过表达的Syn1会通过促进BACE1活性上调来促进APP的淀粉样裂解。其背后的机制尚不清楚,可能是与Syn1能够使BACE1停留在适宜的pH环境中来诱导BACE1的构象向更易底物结合的方向变化[11]。前列腺凋亡反应因子-4(prostate apoptosis response-4, PAR-4)是一种亮氨酸拉链蛋白质,与神经元变性和异常Aβ的产生有关。其C-端与BACE1末端胞质结构域结合,正向调节BACE1活性,促进APP通过淀粉样降解途径裂解。在海马组织中,RNAi介导的PAR-4基因沉默导致BACE1切割APP的活性降低[44]。
3.4 BACE1相互作用蛋白质通过调节BACE1对APP-β位点的切割影响其对APP的裂解
BACE1相互作用蛋白质通过与BACE1结合调节其与底物的相互作用,从而调节BACE1在APP淀粉样降解途径中对APP的切割作用。内质网蛋白3(reticulon3, RTN3)属于RTN家族,定位于内质网,与BACE1的胞内结构域相结合,延长BACE1在内质网中的停留时间,减少BACE1与底物的作用以及向轴突的转运,进而减少BACE1对APP的切割。调查研究显示,RTN3变异与早发型AD有关,但RTN3变异并不直接影响BACE1活性的负调控,可能通过影响RTN3的转录水平影响BACE1[45]。分选蛋白受体A(sorting protein-related receptor, SorLA)又称LRP-II,是一种I型跨膜蛋白质,属于Vps10p受体家族,其肽段尾部与BACE1的胞质结构域结合后,对BACE1裂解APP的过程进行调节,其机制可能与抑制BACE1对β位点的切割有关[46]。
3.5 BACE1相互作用蛋白质通过调节BACE1的降解途径影响其降解水平
BACE1能通过泛素-蛋白酶体途径和溶酶体途径进行降解,BACE1相互作用蛋白质通过与BACE1结合调节BACE1的降解途径,从而调节BACE1的水平。有研究发现,与二磷酸腺苷核糖基化因子(ADP-ribosylation factor, ARF)有相互作用的高尔基体定位的含γ耳ARF结合蛋白(Golgi-localized γ-ear-containing ARF-binding proteins, GGA)的VSH结构域,与BACE1相互作用,调节BACE1在细胞中的运输。GGA3通过与BACE1胞内结构域结合,将BACE1转运到溶酶体来调控其降解, GGA3功能丧失会触发BACE1的积累[47]。GGA1与GGA3协同调节BACE1,GGA1基因沉默能够增强GGA3缺失引起的BACE1升高,而BACE1水平增高时,会显著消耗GGA3,同时也增加了胱天蛋白酶3(caspase-3)介导的GGA1消耗[48]。
BACE1结合蛋白质种类众多,作用机制复杂,除了上述作用机制明确的结合蛋白质外,尚有许多新发现的结合蛋白质作用机制尚未被阐明。例如,脑特异性Ⅱ型膜质(brain-specific type Ⅱ membrane protein, BRI3)可在正常人类和小鼠大脑神经元中与BACE1免疫共沉淀和共定位,两者之间可能的相互作用位点是BRI3的N-端胞质结构域和BACE1的C-端胞质结构域,而BRI3的作用尚未被研究[49]。BACE1胞内结构域尾部的半胱氨酸残基能够与铜原子结合,并与作为超氧化物歧化酶1(superoxide dismutase 1, SOD1)伴侣蛋白质的CCS的N-端结构域有相互作用,抑制细胞中SOD1的活性,参与调节神经的氧化应激。轴突中还存在BACE1和可溶性CCS的共转运[50, 51]。但研究仅停留在与BACE1相互作用的环节,CCS在AD中的作用尚未有研究。
BACE1在神经系统表达广泛,参与生理过程众多,而其作为APP淀粉样降解途径的关键酶,介导了Aβ的生成。BACE1相互作用蛋白质作为BACE1功能调节的重要一环,在AD的发生与疾病进程中占有重要地位,其通过直接结合、间接结合和参与各种细胞信号转导通路等方式直接或间接的对BACE1的转录、翻译、修饰、胞内运输、功能活性的发挥等各个环节进行调控,干预了BACE1的生成、BACE1与APP共定位、APP-β位点的切割、BACE1的降解水平以及Aβ的生成。ATF6、SIRT1、RTN3、SorLA、GGA等BACE1相互作用蛋白质作为AD保护性因素,能够抑制AD的发生减缓疾病的进程,而PS1、AP-2、PAR-4、SYN1等BACE1相互作用蛋白质是AD发病的危险因素,对AD的进展发挥推动作用。深入分析BACE1相互作用蛋白质的作用机制,可以为AD发病机制提供新的理解与思考,也会成为未来AD精准治疗的一个新方向。
4 基于β-分泌酶相互作用蛋白质为靶点的药物研发
在AD早期,Aβ推动疾病进展的作用较后期更加突出,早诊断早治疗尤为重要,而在疾病进程后期去除Aβ斑块不太可能从根本上逆转AD[52]。目前,已经开发了一些肽类和非肽类BACE1抑制剂,但由于BACE1底物众多,从转录起始环节抑制BACE1的表达以及通过抑制BACE1功能活性位点,通常会有严重的不良反应[53]。BACE1抑制剂会引起突触的损伤,BACE1减少会影响突触囊泡的释放,引起记忆异常以及其他神经功能障碍。基于BACE1互作蛋白质靶点的药物研发具有巨大的优势,应当具体研究参与AD发生的BACE1作用机制以及影响因素,将BACE1的药物调控精确地靶向BACE1参与AD发生的具体环节,温和的抑制BACE1功能,在疾病早期不影响记忆的情况下阻断Aβ的生成,突破AD治疗的瓶颈。
目前,已有基于蛋白质相互作用为作用靶点的药物被发现,β-细辛醚(β-asarone)对神经退行性疾病、神经系统肿瘤,以及抑郁症、脑梗死等都有一定的治疗效果[54, 55]。研究表明,该药在促进SYN1的同时抑制了BACE1的表达,减少了Aβ的沉积[56]。氟西汀(fluoxetine)作为选择性5-HT再摄取抑制剂因其不良反应少、口服后吸收良好被广泛应用于抑郁症的治疗中,近期发现,该药物能提高海马组织中突触后密度蛋白95(postsynaptic density protein-95, PSD-95)和SYN-1的水平,减少BACE1的表达[57]。基质金属肽酶13(matrix metallopeptidase 13, MMP13)能够影响BACE1的合成,CL82198是MMP13的抑制剂,通过影响RTK-PI3K信号传导参与MMP13介导的BACE1蛋白调控,降低BACE1蛋白水平[58]。中医药也在AD治疗中展示了其巨大潜力,解毒益智方可通过提高SIRT1 mRNA的含量降低NF-κB mRNA、BACE1 mRNA的表达,从而抑制Aβ的产生而发挥神经保护的作用[59]。虽然在起步阶段,但目前几种通过影响蛋白质相互作用抑制BACE1活性的天然化合物的研究似乎很有希望,但它们的作用机制仍有待确定,需要进一步的探究。
通过蛋白质与BACE1相互作用调节Aβ的生成主要有两个途径:(1)通过调节BACE1互作蛋白质的表达与活性,进而调节BACE1的参与的信号途径,实现对BACE1功能的调节。对BACE1表达与活性无差别的完全抑制会造成严重的不良反应,真正的治疗靶点应该基于抑制BACE1部分活性与表达、对BACE1对APP切割的环节的调控,以及调节BACE1胞内运输,减少BACE1与APP在反式高尔基体与内体的汇合。我们认为,正如RNT3与SorLA等能够调节BACE1对APP切割的互作蛋白质,更有希望成为未来药物研发的突破口。(2)通过抑制或促进蛋白质之间相互作用的亲和力和相互作用强度调节BACE1的功能。蛋白质互作位点缺乏特征、蛋白质之间的非连续性相互作用是药物开发主要的困难,药物对相互作用位点的亲和力和选择性是决定药物性能的2个最重要的属性,因此,增加药物与互作位点的亲和力是未来主攻的方向,抑制BACE1非活性位点是AD治疗的前进方向[60]。
5 问题与展望
AD引起的认知障碍以及神经精神症状严重影响了老年人的生活质量。AD自1906年被提出,已经跨越了1个多世纪,而目前的医疗仍不能治愈该疾病,BACE1作为AD治疗靶点被广泛研究。然而,BACE1抑制剂的研发以及临床试验的现状却不容乐观。近些年,BACE1相互作用蛋白质成为研究热点,但大部分的研究仅介绍了相互作用蛋白质对BACE1的调节作用,并未阐明其作用机制,而作用机制以及互作位点是药物研发的着力点。大部分研究集中揭示了相互作用蛋白质对BACE1细胞内转运的调节,而对BACE1构象活性的调节鲜有研究。未来关于BACE1相互作用蛋白质的研究应当继续深入,该领域需要充分揭示BACE1互作蛋白质的生理功能,研究成果将为AD的预防、治疗以及新型药物研发提供新的思路。
猜你喜欢
农业技术与装备(2022年6期)2022-08-17
上海金属(2021年6期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
生物信息学(2020年1期)2020-05-16
广州大学学报(自然科学版)(2019年1期)2019-05-07
生物学通报(2019年3期)2019-02-17
电脑知识与技术(2018年19期)2018-11-01
西安工程大学学报(2016年6期)2017-01-15
天津科技大学学报(2016年1期)2016-02-28
中国粮油学报(2016年1期)2016-02-06
