
长链烯烃氢甲酰化反应催化剂研究进展
2023-02-21 07:25:22燕子红郑源松苗志伟
石油化工 2023年1期
燕子红,郑源松,蔡 岩,苗志伟
(1.喀什大学 化学与环境科学学院,新疆 喀什 844006;2.天津国际生物医药联合研究院,天津 300457;3.南开大学 化学学院 元素有机化学国家重点实验室,天津 300071)
烯烃氢甲酰化反应是化工生产中应用广泛的有机合成反应之一,该反应为烯烃在催化剂作用下与合成气(即氢气与CO 混合气)反应,生成比烯烃多一个碳原子的醛类化合物,反应过程具有良好的原子经济性[1]。2019 年,全球通过烯烃氢甲酰化反应生产的醛类化合物已超过100 Mt[2]。醛类化合物可进一步加工成醇、酯、醚、羧酸以及胺等化合物,这些产品在增塑剂、表面活性剂、药物合成、洗涤剂和香料等方面广泛应用,因此烯烃氢甲酰化反应在工业上具有重要的应用价值[3]。
本文综述了长链烯烃(碳数大于5)氢甲酰化反应的研究进展,介绍了长链烯烃氢甲酰化反应催化剂所用膦配体的种类及合成方法。
1 长链烯烃氢甲酰化反应催化剂所用膦配体
长链烯烃氢甲酰化反应催化剂的选择是反应顺利进行的关键,其中,膦配体的电子效应和空间效应对催化剂的活性与区域选择性具有重要影响。
1.1 单齿膦配体
典型的单齿膦配体结构见图1。单齿膦配体的结构相对简单,其中,三苯基膦(配体1)是较早实现工业应用的膦配体。膦配体改性的铑催化剂可以使烯烃氢甲酰化反应在温和条件下进行,得到的醛产物中正构醛与异构醛的摩尔比(正异比)提高。Smith 课题组[4]分别使用配体1 和配体2 改性羰基铑催化剂,在1-辛烯氢甲酰化反应中对正壬醛的区域选择性均超过80%。Maura 等[5]使用配体3 改性铑催化剂,在1-辛烯氢甲酰化反应中的正异比达2.20。Deshmukh 等[6]使用配体4 改性铑催化剂,在1-辛烯氢甲酰化反应中对正壬醛的区域选择性超过60%。
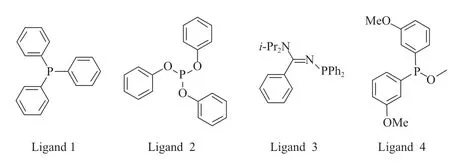
图1 典型单齿膦配体的结构Fig.1 Structure of typical monodentate phosphine ligand.
Fleming 等[7]认为,与三苯基膦相比,在单齿亚磷酸酯类配体中,与磷原子相连的苯氧基是吸电子基团,而处于缺电子状态下的磷原子有利于稳定低氧化态的过渡金属,能够促进CO 与过渡金属中心解离。考虑到膦配体的电子效应,亚磷酸酯类膦配体(结构见图2)逐渐受到广泛关注。Selent 等[8]采用配体5 改性铑催化剂,在1-辛烯氢甲酰化反应中的正异比达1.57。Dabbawala 等[9]采用配体6 改性铑催化剂,在1-己烯氢甲酰化反应中对正构醛的选择性超过70%。Rooy 等[10]发现,大位阻的亚磷酸酯类配体7 与金属铑配位,在催化1-辛烯以及1-十二烯的氢甲酰化反应中表现出较好的区域选择性,对正构醛的选择性分别为66%和63%。
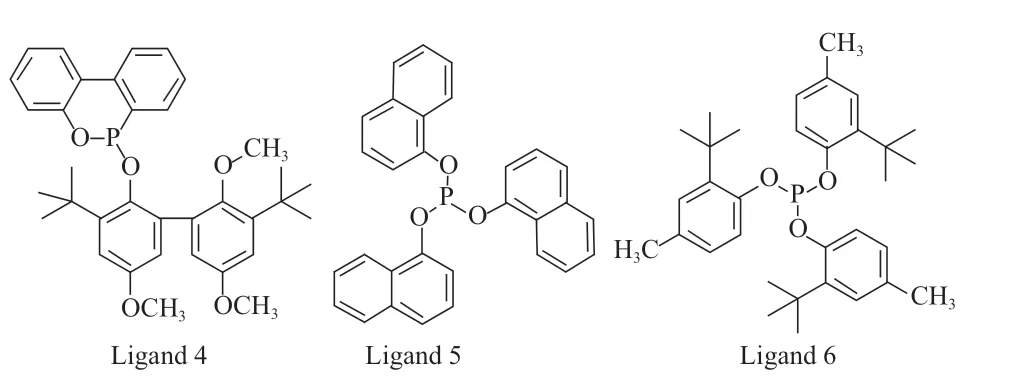
图2 亚磷酸酯类膦配体的分子结构Fig.2 Molecular structure of phosphite ligands.
单齿亚磷酸酯类配体在烯烃氢甲酰化反应中具有良好的催化活性,然而,该类配体不稳定,易水解失活,阻碍了它在工业上的广泛应用[11]。通过改变膦配体的分子结构能够有效改善催化剂活性和区域选择性,因此,基于磷杂苯结构的膦配体(结构见图3)逐渐受到研究者的关注。Breit等[12]将含磷杂苯结构的膦配体与乙酰丙酮二羰基铑(Rh(CO)2(acac))反应制成络合物,用于1-辛烯氢甲酰化反应,采用配体8 时单位时间内生成目标产物的转化量(TOF)达到2 801 h-1,正异比为2.00;采用配体9 时的TOF 为2 443 h-1,正异比为1.80;采用配体10 时的TOF 达到5 751 h-1,正异比高达3.30;采用配体11 时的TOF 为1 951 h-1,正异比为2.00。配体10 制备的催化剂在反应中表现出较高的催化活性和区域选择性。实验结果表明,催化剂的活性和区域选择性除了受电子效应影响外,磷原子邻位的空间位阻也是影响烯烃氢甲酰化反应正异比的重要因素。
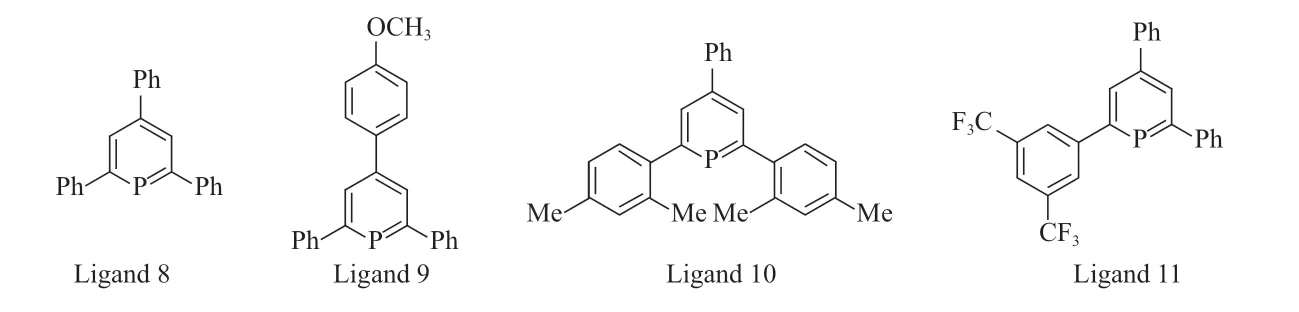
图3 含磷杂苯结构的膦配体的结构Fig.3 Ligand structure of phosphorus containing heterobenzene.
1.2 双齿膦配体
与单齿膦配体相比,双齿膦配体具有螯合效应,更容易与过渡金属配位,形成至少含有两个磷原子中心的中间体,从而促进反应进行。1992 年,Casey 等[13]研究了双齿膦配体的分子结构,对一些典型双齿膦配体(结构见图4)对1-己烯氢甲酰化反应区域选择性的影响进行了比较,结果见表1。由表1 可知,在与二羰基铑络合催化1-己烯的氢甲酰化反应中,配体12 的催化活性和区域选择性都非常高,正异比为66.5。Casey 等[14]以不同膦配体与金属配位形成的P—M—P(M 表示金属)之间的键角(咬角)来评估双齿膦配体的空间效应对催化剂的影响。Casey 等认为,配体12 在烯烃氢甲酰化反应中选择性好的原因是它与配位金属能够形成较大的咬角。虽然配体14 的咬角大,但正异比很低,这是由于未能分离出配体14 的单体金属配合物,猜测可能是因为配体14 不能与铑形成螯合的配合物所致。
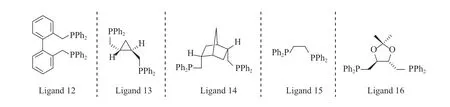
图4 典型的双齿膦配体结构Fig.4 Typical bidentate phosphine ligand structure.
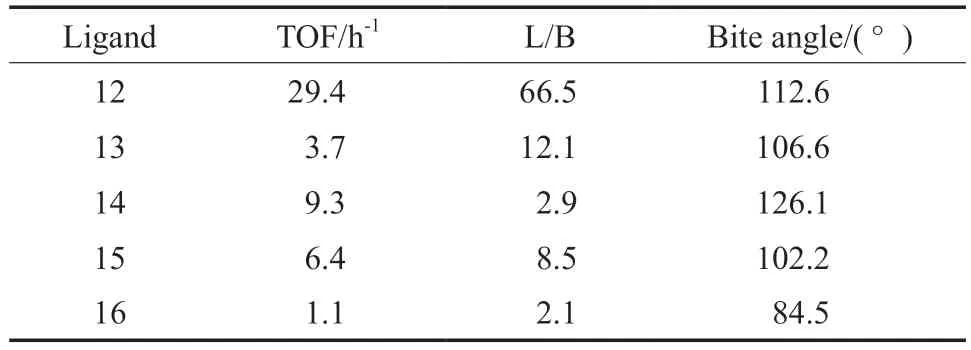
表1 铑-双膦配体催化剂催化1-己烯氢甲酰化反应结果Table 1 Results of hydroformylation of 1-hexene catalyzed by rhodium diphosphine ligands
Xantphos 系列配体是具有刚性杂蒽结构的双(齿)膦配体。van der Veen 等[15]报道了具有相似结构的Xantphos 系列配体(结构见图5),及其在1-辛烯氢甲酰化反应中的结果(见表2)。由表2 可知,Xantphos 系列配体与Rh(CO)2(acac)络合催化1-辛烯氢甲酰化反应时,对正构醛的选择性与催化剂配体形成的咬角成正比。当P—Rh—P 之间的咬角从配体17 的102°提高到配体25 的120°时,正壬醛的选择性从88%提高到97%。
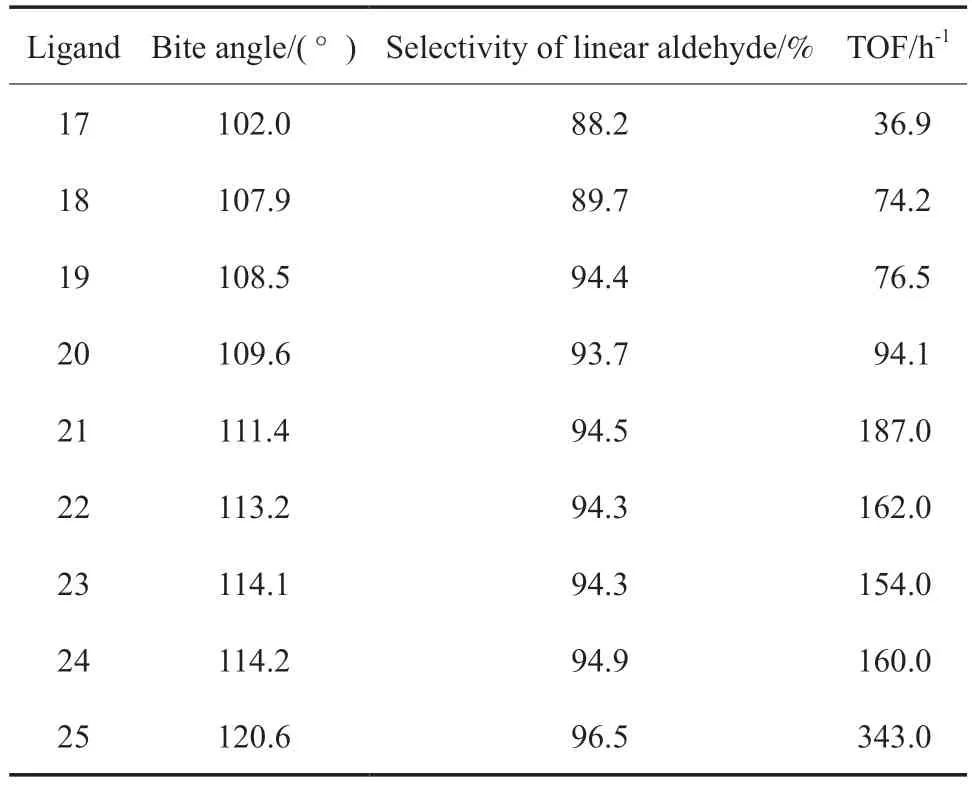
表2 Xantphos 系列配体用于1-辛烯氢甲酰化反应结果Table 2 Results of 1-octene hydroformylation with Xanthos ligands
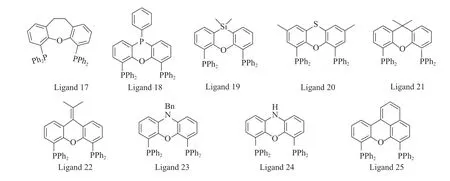
图5 Xantphos 系列配体的结构Fig.5 Structures of Xanthos series ligands.
在含有N—P 键结构的催化剂配体方面,Jia 等[16]报道了双齿亚磷酰胺配体26 与Rh(CO)2(acac)络合催化1-己烯氢甲酰化反应的结果,正异比达15.70,催化剂有着优异的区域选择性。van der Slot 等[17]分别采用配体26 和配体27 与金属铑络合,在1-辛烯氢甲酰化反应中,含有吡咯基的亚磷酰胺配体26 对正构醛的选择性达到92%,苯基取代的配体27 对正构醛的选择性为85%。由此可见,吡咯基团与苯基的电子效应差异是影响烯烃氢甲酰化反应选择性的重要因素。双齿亚磷酰胺配体26 和双齿亚膦酸酯配体27 的结构见图6。
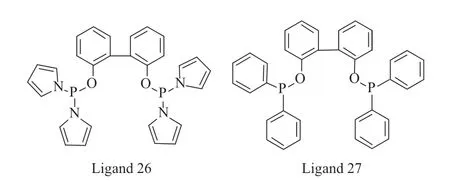
图6 双齿亚磷酰胺配体26 和双齿亚膦酸酯配体27 的结构Fig.6 Structures of bidentate phosphorus amidite and phosphinite ligands.
1.3 多齿膦配体
与单齿膦配体相比,双齿膦配体与金属的螯合能力更强,减少了反应过程中膦配体的用量,而多齿膦配体一般体积较大,有更多的磷原子可以与金属螯合,并且分子内游离的磷原子可以有效增加金属中心附近的局部磷浓度,使催化剂配体的螯合能力进一步增强。研究结果表明,双齿亚磷酰胺配体26 产生高区域选择性的原因是吡咯基团的强吸电子性质。因此,Yan 等[18]设计合成了具有吡咯基对称结构的四齿膦配体28。在配体28 与铑络合催化1-辛烯的氢甲酰化反应中,正异比高达372.00。而双齿膦配体26 在同样的反应条件下催化1-辛烯氢甲酰化反应的正异比为74.10。在磷酸酯类配体方面,Zhang 等[19]采用配体29 与Rh(CO)2(acac)络合催化1-辛烯氢甲酰化反应,正异比达65.00。同样的反应条件下,四齿膦配体对正构醛选择性总是优于相应的双齿膦配体。四齿膦配体结构见图7。
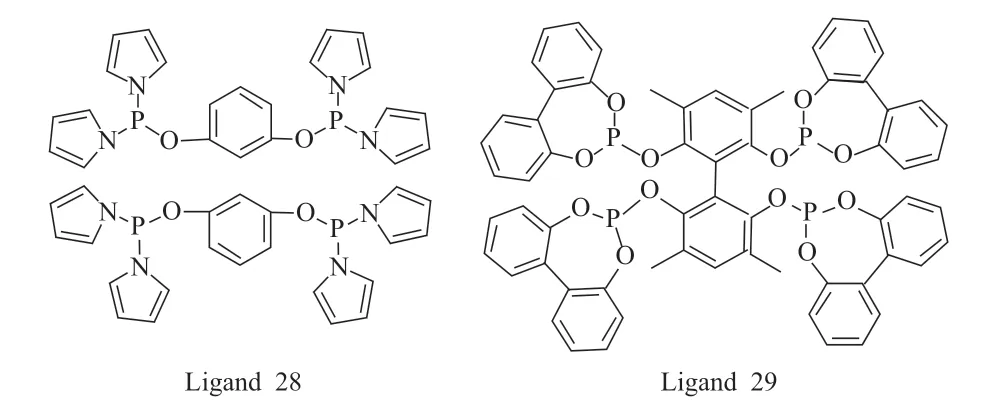
图7 含O—P 键的四齿膦配体结构Fig.7 Structures of tetraphosphorus ligands containing O—P bond.
2010 年,Yu 等[20]设计合成了类似配体12 结构的基于联苯骨架的四齿膦配体(结构见图8),该配体30 与Rh(CO)2(acac)络合催化1-辛 烯的氢甲酰化反应,反应温度为100 ℃时,正异比为50.50;同样条件下采用配体12 时,正异比为45.20。当其他条件不变,将反应温度升高到140 ℃,采用配体30 时,正异比为45.20,而采用配体12时正异比仅为2.40。与相应的双齿膦配体相比,四齿膦配体拥有更好的区域选择性及稳定性。

图8 基于联苯骨架的四齿膦配体结构Fig.8 Structure of tetraphosphorus ligand based on a biphenyl backbone.
2 长烯烃氢甲酰化反应催化剂所用膦配体的合成
2.1 单齿膦配体的合成
2018 年,Xu 等[21]成功地将介孔硅材料MCM-41 和三齿氮钯(0)的复合物应用于芳基碘苯(31)与二苯基膦(32)的交叉偶联反应,在惰性气体保护下制备了配体1,收率为86%。这种介孔硅材料复合催化剂可以至少重复使用7 次,降低了生产成本,同时也避免了单独使用过渡金属铜、钯、镍催化可能造成的重金属污染问题。配体1 的合成见图9。
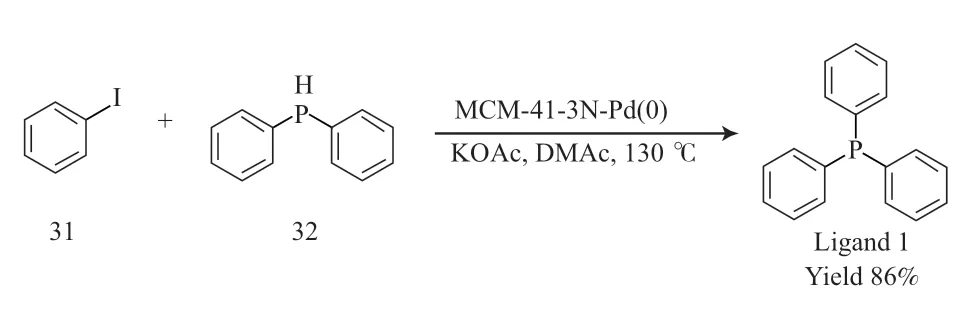
图9 配体1 的合成Fig.9 Synthesis pathway of ligand 1.
2021 年,Zhang 等[22]以白磷(33)与苯酚(34)为原料,在二苯基二硒催化下一步合成了配体2,收率为88%。该反应提供了一种无卤素、无过渡金属催化合成配体2 的新路线,避免了传统工艺中氯气和三氯化磷的使用,以及大量HCl 和金属卤化物等“三废”的产生。配体2 的合成见图10。
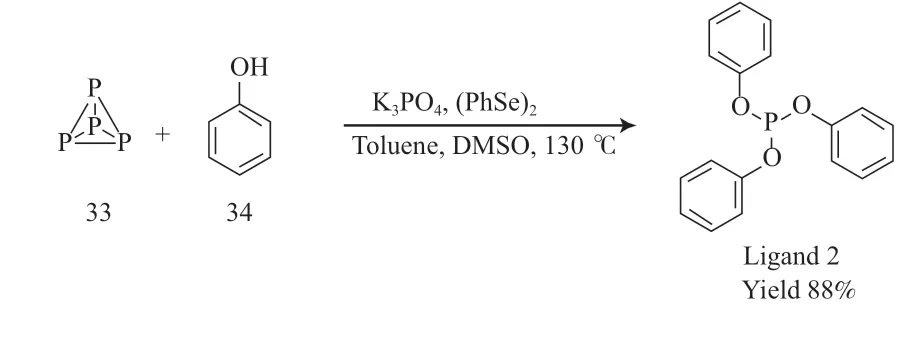
图10 配体2 的合成Fig.10 Synthesis pathway of phosphite ligand 2.
配体8 是1966 年由Märkl[23]首次合成的,以2,4,6-三苯基吡喃四氟硼酸盐(35)为原料,与三(羟甲基)膦在吡啶中回流,最终得到配体8,收率为24%~30%。2016 年,Rigo 等[24]改进了配体8 的合成方法,以2,4,6-三苯基吡喃四氟硼酸盐(35)为原料,双(三甲基硅基)膦锂盐为磷试剂,在四氢呋喃中回流,制备的配体8 的收率为76%,与原有合成方法相比,收率大幅提升。配体8 的合成见图11。
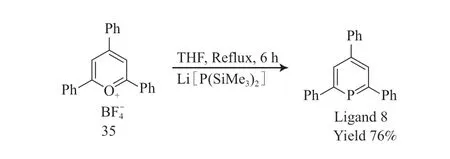
图11 配体8 的合成Fig.11 Synthesis pathway of phosphorus containing heterobenzene ligand 8.
2.2 双齿膦配体的合成
Xantphos 系列配体17~25 都是通过选择合适的杂环化合物作为起始原料合成的。以双(2-(二苯基膦基)苯基)醚(36)为原料,利用三氟甲磺酸催化它与丙酮的缩合反应,制备了配体21,收率为95%[25]。该反应具有路线短、后处理简单、原料较为便宜的优点。配体21 的合成见图12。
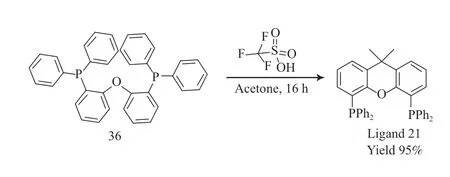
图12 配体21 的合成Fig.12 Synthesis pathway of ligands 21.
2021 年,Jiang 等[26]采用2,2'-二羟基-1,1'-联苯(37)为原料合成了双齿亚磷酰胺配体26。以四氢呋喃为溶剂,使用过量的三乙胺为缚酸剂,二吡咯氯化磷与2,2'-二羟基-1,1'-联苯(37)发生亲核取代反应合成配体26,收率为68%。后处理采用较为简便的重结晶方法进行产物提纯。配体26 的合成见图13。
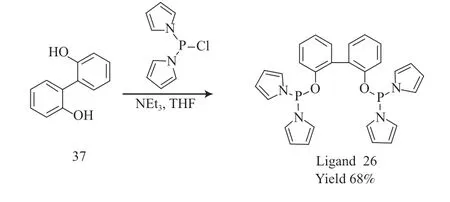
图13 配体26 的合成Fig.13 Synthesis pathway of bisphosphite ligand 26.
2.3 多齿膦配体的合成
Lindsten 等[27-28]以廉价的1,3-二甲氧基苯(38)为原料,采用正丁基锂催化苯环的碘代反应,得到的2-碘-1,3-二甲氧基苯(39)通过Ullmann偶联反应发生自缩合,得到2,2',6,6'-四甲氧基联苯(40),收率为90%。三溴化硼是Lewis 酸,用于2,2',6,6'-四甲氧基联苯(40)的脱甲基,得到的2,2',6,6'-四羟基-1,1'-联苯(41)的收率为80%。在三乙胺存在下,使用二吡咯氯化磷与2,2',6,6'-四羟基-1,1'-联苯(41)反应得到四齿膦配体28,收率为70%。配体28 的合成见图14。
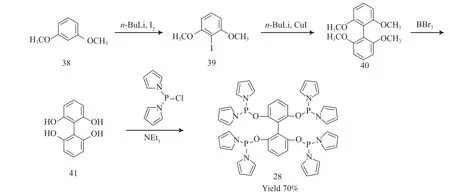
图14 配体28 的合成Fig.14 Synthesis pathway of amide tetraphosphorus ligand 28.
2010 年,Yu 等[20]以芘(42)为原料通过臭氧化开环反应,在碘化钠存在下进行偶联得到中间体(43)。利用硼氢化钠还原2,2',6,6'-四甲醛-1,1'-联苯(43)得到2,2',6,6'-四(羟甲基)-1,1'-联苯(44),再与三溴化磷反应以高收率制得四溴代中间体2,2',6,6'-四(溴甲基)-1,1'-联苯(45)。因为中间体(45)与二苯基膦反应会产生复杂的混合体系,所以首先在N,N-二甲基甲酰胺中用氯化锂将中间体(45)转化为反应活性较差的2,2',6,6'-四(氯甲基)-1,1'-联苯(46),再进一步与二苯基膦的锂盐反应生成产物(47)。考虑到最终产物配体30 对空气敏感,因此先利用硼烷的四氢呋喃溶液将它保护起来,经纯化后用1,4-二氮杂环[2.2.2]辛烷脱保护,得到配体30,收率为64%~79%,合成示意见图15中的式(1)。还可以利用氯化亚砜与2,2',6,6'-四(羟甲基)-1,1'-联苯(44)反应,以100%的收率一步转化为2,2',6,6'-四(氯甲基)-1,1'-联苯(46),进一步在锂的四氢呋喃溶液中与二苯基氯化膦反应,以90%的收率得到配体30[29],合成示意图见图15 中的式(2)。该合成策略减少了合成步骤,提高了最终产物的收率。
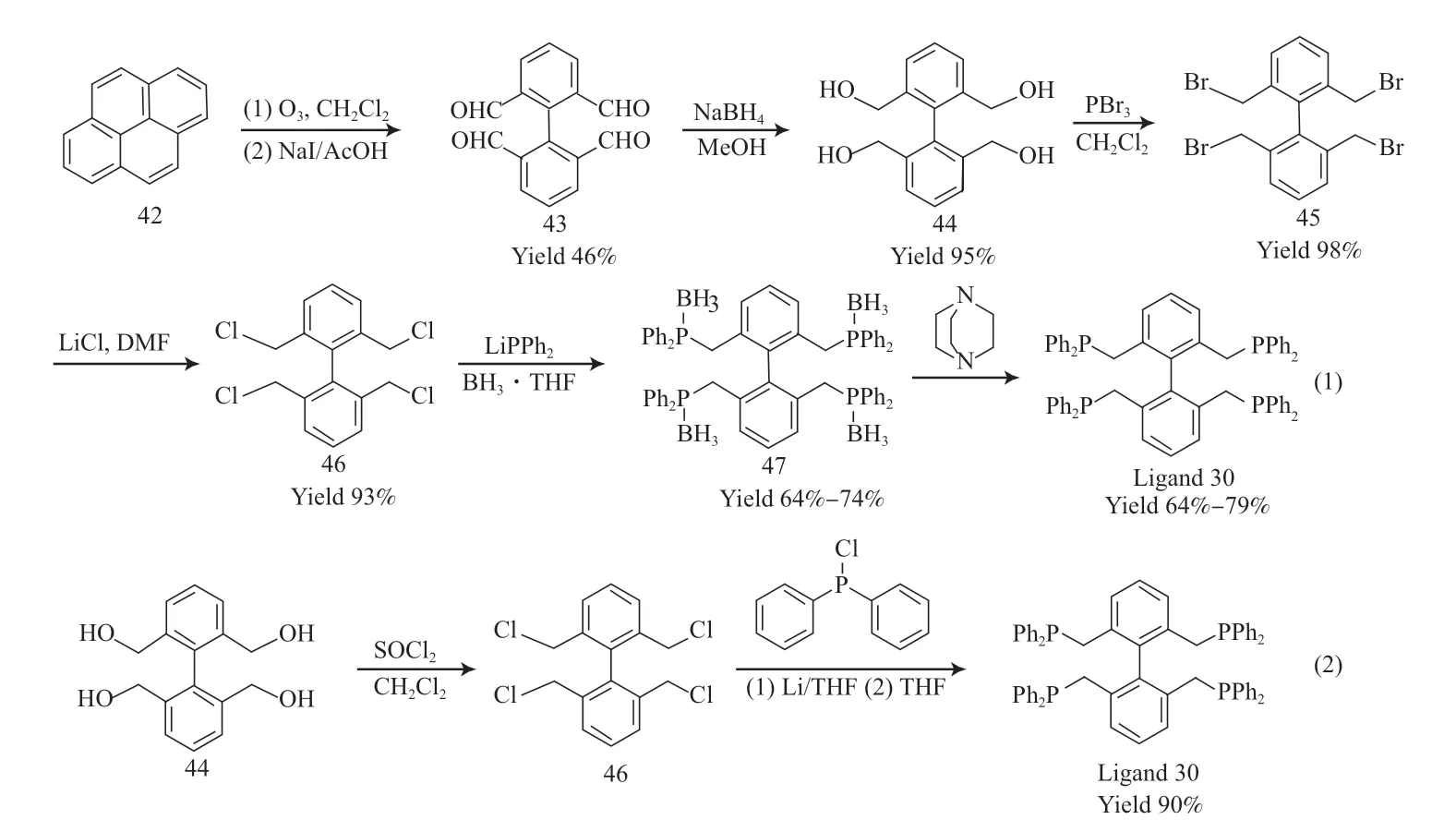
图15 配体30 的合成Fig.15 Synthesis pathway of tetradentate phosphine ligand 30.
3 金属催化剂的制备
Rh(CO)2(acac)是烯烃氢甲酰化反应的催化剂前体,在氢甲酰化反应条件下,Rh(CO)2(acac)首先与膦配体结合,形成具有催化活性的HRh(CO)(L)2络合物(L 表示膦配体)。Rh(CO)2(acac)的早期合成方法[30]是以RhCl3·xH2O 为起始原料,通入CO 得到[Rh(CO)2C1]2,在BaCO3催化下与乙酰丙酮反应7 d 后,得到Rh(CO)2(acac),收率为85%,合成示意见图16 中的式(1)。2020年,中海油天津化工研究设计院有限公司[31]以Rh2O3·xH2O 为起始原料,在乙酸催化下与乙酰丙酮反应2 h,得到Rh(CO)2(acac),收率为99%,合成示意见图16 中的式(2)。该合成方法提高了最终产物的收率,大幅缩短了反应时间,提高了经济效益。
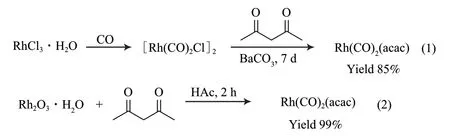
图16 金属催化剂的合成Fig.16 Synthesis of metal catalyst.
4 结语
烯烃氢甲酰化反应已经发展成为重要的醛类化合物生产工艺之一。利用铑催化剂均相催化长链烯烃(碳数大于5)氢甲酰化反应的技术也日臻完善,包括单齿膦配体、双齿膦配体和多齿膦配体在内的各种类型的膦配体已经被开发并应用于工业生产。尽管目前烯烃氢甲酰化反应的生产工艺水平很高,但在长链烯烃氢甲酰化反应研究方面仍存在许多不足。未来围绕长链烯烃氢甲酰化反应应主要开展以下研究工作:1)开发催化活性高、稳定性好、区域选择性优异的膦配体;2)开发新型均相催化生产工艺和催化剂分离及循环套用技术,使生产工艺更简单、更环保、更经济;3)开发低压长链烯烃氢甲酰化反应生产工艺,提高生产过程的安全性。未来随着长链烯烃氢甲酰化反应理论研究的深入和生产水平的不断提高,该领域一定会迎来更大的发展。
猜你喜欢
石油石化节能(2023年1期)2023-02-12 04:23:04
中国饲料(2021年17期)2021-11-02 08:15:14
广东饲料(2016年5期)2016-12-01 03:43:22
癌变·畸变·突变(2016年3期)2016-02-27 06:15:25
合成化学(2015年2期)2016-01-17 09:03:13
合成化学(2015年10期)2016-01-17 08:56:37
中国塑料(2015年6期)2015-11-13 03:02:48
化工进展(2015年3期)2015-11-11 09:08:25
橡胶工业(2015年6期)2015-07-29 09:20:30
化学分析计量(2015年4期)2015-03-23 16:47:34
