
基于深度学习原子特征表示方法的Janus过渡金属硫化物带隙预测*
2023-02-18 06:39:00孙涛袁健美
物理学报 2023年2期
孙涛 袁健美†
1) (湘潭大学数学与计算科学学院,湘潭 411105)
2) (科学工程计算与数值仿真湖南省重点实验室,湘潭 411105)
随着人工智能的发展,机器学习在材料计算中的应用越来越广泛.将机器学习应用到材料性质预测等任务中首要实现的是获得有效的材料特征表示.本文采用一种原子特征表示方法,研究一种低维、密集的分布式原子特征向量,并用于材料带隙预测任务.按照材料化学式中原子种类和原子个数,使用Transformer 编码器作为模型结构,通过训练大量的材料化学式数据,从而提取参与训练元素的特征.利用该方法预测Janus结构过渡金属硫族化合物MXY (M 代表过渡金属,X,Y 是不同硫族元素)二维材料带隙.基于深度学习得到的原子特征向量比传统的Magpie 方法和Atom2Vec 方法的预测平均绝对误差更小.可视化分析和材料性质预测数值实验表明,本文提出的基于深度学习提取的原子特征表示方法,可以有效表征材料特征,并且应用到材料带隙预测任务中.
1 引言
传统的材料科学研究通常需要经过大量的计算得到材料的目标属性,这通常会消耗大量时间和资源.随着人工智能的快速发展,深度学习技术已经被广泛应用在图像识别[1]、目标检测[2]和自然语言处理[3]等领域.深度学习技术不需要了解从特征空间到目标值的具体函数关系,而是通过训练大量的数据,得到了一组神经网络权值来构建从特征空间到目标值的映射关系.近年来,深度学习技术被应用到材料的发现和设计、材料性质预测[4,5]等方面.Hu 等[6]通过在OQMD 数据库上训练了一个WGAN模型,利用训练好的鉴别器模型,得到了一种材料表征方法,并且通过在公共 数据集上对材料的带隙、形成能和临界温度进行预测验证了其有效性.Chen 等[7]利用图神经网络建立MEGNet 来预测分子和晶体的性质,在QM9 数据集上预测了13 个目标性质,其中11 个性质优于之前的预测效果.Li 等[8]提出了一种结合卷积神经网络和长短期记忆神经网络的混合神经网络,用于超导体的临界温度预测.此外,深度学习技术也被用于分子动力学模拟中,如鄂维南课题组[9]提出了一种基于神经网络的分子动力学模拟方案,克服了与辅助量(如对称函数或库仑矩阵)相关的限制,通过构造深度学习原子势来描述原子周围的环境.从大量的材料数据中学习潜在的物理规律,可以用于材料性质的预测,这通常会节省大量的时间和资源.
机器学习模型最重要的两个方面是数据表示和学习算法.在材料性质预测任务中,机器学习模型的数据表示就是确定材料的特征描述符.在之前的研究工作中,特征描述符的确定通常是先根据预测材料的特性,按经验从已有的原子特征或者材料结构特征中初步选择一些特征,然后再利用特征选择方法通过尝试和试错逐步确定最终的特征[10].为了得到材料的通用特征描述符,越来越多的材料表征方法被不断提出.Zhou 等[11]提出Atom2Vec方法得到原子向量表示,用材料数据建立原子-环境矩阵,通过奇异值分解降维方法得到原子20 维向量表示.Li 等[8]利用Atom2Vec 方法得到的原子向量构建了超导体材料的稀疏矩阵数据表示.Calfa 等[12]提出One-hot 方法得到二元金属氧化物和晶体材料数据表示,利用核回归预测了金属氧化物的电子性质和晶体的弹性性质.One-hot 方法可以简单地为其他类型材料提供数据表示,有很多学者利用One-hot 方法得到的材料表征作为材料的数据表示,进行下一步机器学习任务.例如Hu 等[6]通过One-hot 方法将OQMD 数据库中材料表示成一个稀疏矩阵.Ward 等[13]提出Magpie 方法表征材料,通过开发一组基于组合的通用属性集,得到材料的一维向量数据表示.在文献[14]中,使用Magpie 方法表征材料,采用支持向量机模型建立了预测无机固体的带隙机器学习模型.
在之前的材料信息学研究中,材料的数据表示方法通常为以下两种: 一是先得到原子的数据表示(Atom2Vec),然后通过材料化学式中原子的组成和数量拼接原子向量得到材料的数据表示;另一种是直接通过材料化学式或材料的物理化学性质数据,得到材料的数据表示,例如One-hot 方法.本文利用开放量子材料数据库的大量材料数据,以自监督的方式训练Transformer 编码器模型,提取嵌入层参数得到原子特征向量.然后,通过对主族元素原子的特征向量进行聚类分析,实现了提取的原子特征向量可以区分元素的类别;对主族元素原子特征向量的主成分进行降维分析可以看到,原子特征向量在第一主成分上的投影基本反映了该元素对应的最外层电子数;最后,将其应用在Janus 结构的过渡金属硫族化合物二维材料带隙的预测任务中,验证了原子特征向量在材料预测任务中的有效性.
2 原子特征提取模型介绍
2.1 模型结构
该模型是一种预训练的机器学习模型.用机器学习模型解决材料问题时,常常面临数据量不足的问题.如果直接用该数据进行下游任务(材料性质预测等),训练效果可能一般.在材料性质预测任务中,模型输入特征一般包括原子特征和材料结构特征[15].本模型的主要作用就是在大量的材料数据中提取原子特征,为用机器学习模型进行材料性质预测等任务得到可靠的输入.
本模型基于性质相似的原子可以和同样的原子形成结构和性质相似的化合物的观点.例如氟和氯是同族原子,都可以和氢以1∶1 的比例结合形成氟化氢(HF)和氯化氢(HCl).
在自然语言处理任务中,Transformer 是一种经典的神经网络框架[16].Transformer 包括编码器和解码器两部分.本模型结构使用Transformer 的编码器部分,如图1 所示,图中蓝色矩形代表原子向量.每个Block 输入原子向量和输出原子向量的个数是相同的,因此可以叠加多个Block.在样本输入Block 进行训练前,首先会生成一个原子词汇表,原子词汇表包含了训练数据中全部的原子和特殊符号.对每一个材料化学式中的原子使用One-hot编码得到一个原子词汇表长度的向量.通过一个神经网络嵌入层,让原子的向量维度从原子词汇表长度减少到嵌入层神经元个数,在将其输入Transformer 的Block 中进行训练.在损失函数的控制下,通过反向传播算法更新模型中各个节点的参数,待损失函数值趋于稳定,提取了模型前面的嵌入层参数,即原子词汇表中每个原子的特征表示.
每个Block 内部结构如图1 右图所示,每个Block 的输出向量都由输入Block 的向量经过同样的处理方式得到输出向量.以图1 左图从下往上第一个Block 的橙色输出向量为例,介绍每个Block内部机制(橙色向量仅仅用于介绍Block 内部机制,和蓝色向量都代表原子特征向量).输入Block的橙色向量首先会经过注意力机制得到一个包含其他输入向量信息的输出向量,然后将输入注意力机制的橙色原子向量和该输出向量相加,得到的新向量经过层归一化操作,输入到全连接层中得到输出向量;该向量和输入全连接层的向量相加,再经过层归一化操作得到Block 的对应输出向量.图1右图中仅展示一个Block 的输出向量,对于其他输出向量,由输入向量经过同样的计算过程得到.

图1 Transformer 编码器结构Fig.1.The Transformer encoder structure.
图1 中注意力机制由一个多头注意力机制组成,如图2 所示.对于输入注意力机制的全部向量组成的矩阵M ∈Rd×f(d为向量个数,f为向量维度),分别与可训练矩阵Q ∈Rf×f,K ∈Rf×f,V ∈Rf×e(e为向量线性变换到v矩阵的维度)相乘得到q ∈Rd×f,k ∈Rd×f,v ∈Rd×e,公式如下:
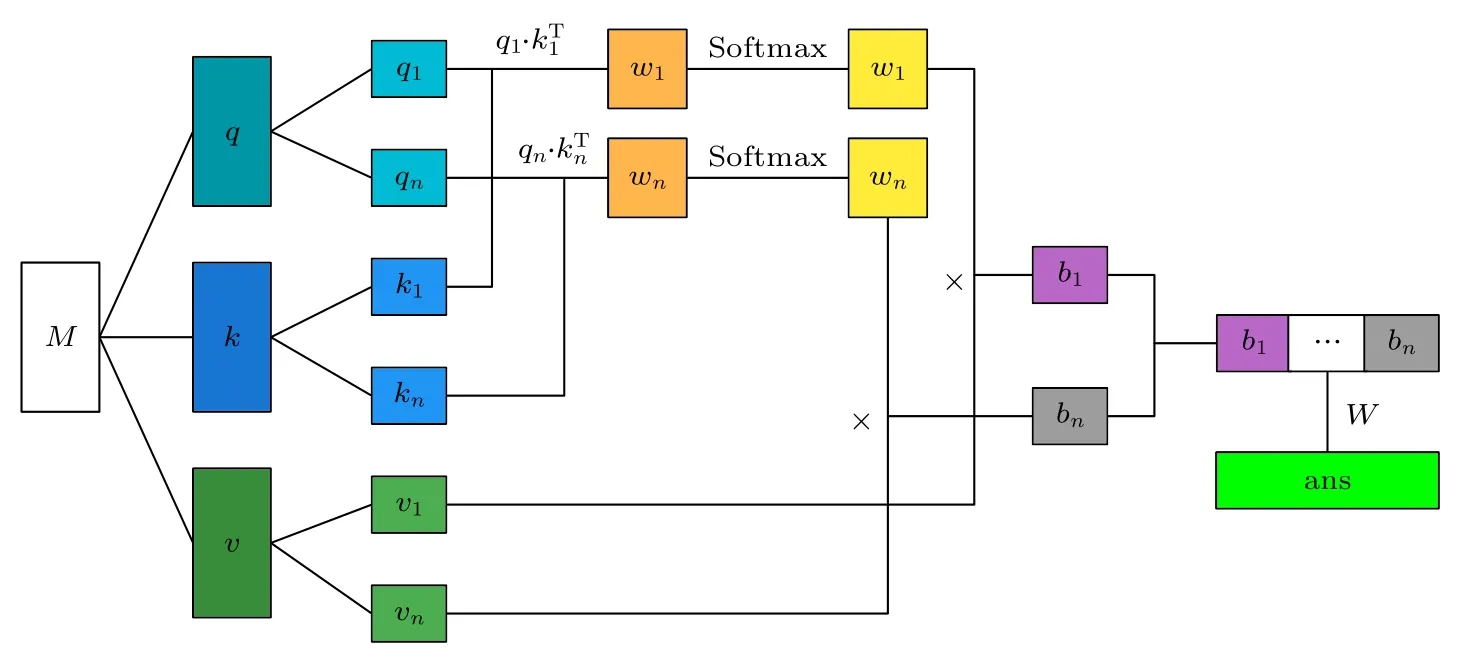
图2 多头注意机构模块结构[16]Fig.2.Multi-attention mechanism module structure[16].

注意力机制输出矩阵的计算公式如下:

多头注意力机制每个hea d 对q,k,v分别进行线性变换,对每个head 执行注意力函数.将 headi得到的结果向量记为bi,所有head 得到的结果拼接起来再进行线性变换,得到最终多头注意力机制的输出向量ans.

2.2 模型训练方法
如图3 中示例输入样本所示,将输入样本表示成材料化学式1、分隔符、材料化学式2、样本标签4 部分.受到自然语言处理领域表现出良好性能的Bert 模型[3]的启发,使用的训练方法有两种,如图3 所示.第一种训练方法是随机遮盖掉一条输入样本15%的原子,让模型来预测这些被遮盖掉的原子.如果某个原子被遮盖,在训练时该原子位置的输入有3 种情况: 有80%的概率替换成特殊字符[MASK],有10%的概率随机替换成一个原子,另外10%概率替换为原子本身.被遮盖掉的原子对应的输出向量会经过一个线性多分类器,用softmax 函数得到预测结果的概率分布,然后与该原子的真实标签用交叉熵损失函数计算损失值,得到 l osslm(原子真实标签可以在模型训练过程中遮盖原子操作时产生).这样的训练方式将具有相似性质的原子得到相似的原子向量.

图3 模型训练示意图Fig.3.Model training diagram.
第二种训练方法是对化合物做类别预测.若两个材料化学式中包含的元素属于化学元素周期表中同样的族,则将两个材料认为是同一类.若一条样本中两个材料是同一类别,则该样本的标签为1;反之,若一条样本中两个材料不是同一类别,则该样本标签为0.在实际训练时,在每条样本开始会加入一个特殊字符[CLS].由于模型中加入了注意力机制,该特殊字符在在参与训练时考虑到了整个样本,所以对特殊字符[CLS]输出结果做一个二分类任务,判断同一条样本中的两个材料是否属于同一类别.该二分类任务的损失函数也是交叉熵损失函数,通过计算损失值得到 l osslabel.
在训练过程中,以上两种训练方法同时进行,然后对目标模型做如下优化:
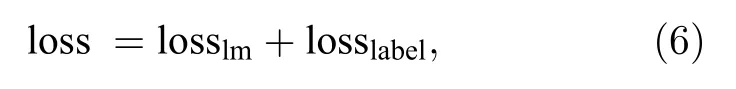
通过反向传播算法,对模型中的参数进行更新,来减少总损失值,直到总损失值收敛.
3 数值实验和结果分析
模型需要材料化学式数据作为输入数据进行训练,使用开放量子材料数据库[17](OQMD)的数据来训练模型.开放量子材料数据库包含大量由密度泛函理论计算的晶体结构数据,从OQMD 数据库中提取561888 个材料的化学式,按照模型输入样本格式,将其重组成560130 条样本用于训练模型.
模型基于Pytorch[18]框架,利用其自动微分和GPU 加速计算动态张量,同时保持较快的计算速度.将提取的样本中包含的所有元素组成一个原子词汇表,为了得到原子词汇表中的所有元素的分布式向量表示,需要先确定向量维度.由于得到的向量应具备密集、低维的特点,所以向量维度不能高于所有元素的个数,但是向量维度太低可能不能包含学习到的全部信息,所以将嵌入层神经元个数调整为16,最终也将得到元素的16 维原子向量.Transformer 编码器模型已经在自然语言处理领域广泛应用,其中一个重要的模型应用就是BERT模型[3].本文深度神经网络的参数设置参考BERT模型参数,并在实际训练过程中进行参数微调.最终将模型中Transformer 的Block 数设为8,多头注意力机制head 数目设为8,训练80 代.
以主族元素为例,分析提取的原子向量代表的物理化学性质.对参与训练的34 个主族元素进行了基于余弦距离(见 (7)式)的层次聚类,聚类结果如图4 所示.
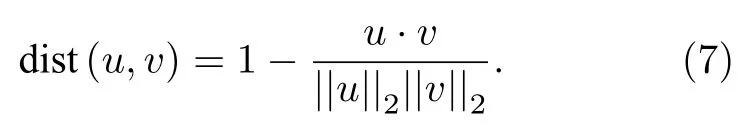
在图4 中,将相似的原子特征向量用红色矩形标出.可以看到,第I 主族碱金属元素(Li,Na,K,Rb,Cs)和II 主族碱土元素(Be,Mg,Ca,Sr,Ba)全部元素被分为一组;第 III 主族金属元素(Al,Ga,In,Tl)被分为一组;非金属元素(H,C,N,P,O,S,Se,F,Cl,Br,I)除氧元素为都被分为一组,包含典型的非金属元素——卤素;类金属元素(B,Si,Ge,As,Sb,Te)中,元素B,Ge,As,Sb 被分为一组,同组还有第IV 主族金属元素(Sn,Pb),第V主族金属元素(Bi),因为同族元素具有相似的化学性质,这也许是模型更倾向于学习Sn,Pb,Bi 和同族元素的化学性质而不是元素类别.除此之外,利用深度学习得到的原子特征向量也提取了部分原子序数相邻的元素的关系.例如Al 和Si 的原子序数分别为13 和14,由图4 可以看出,模型提取的原子特征向量也极为相似.
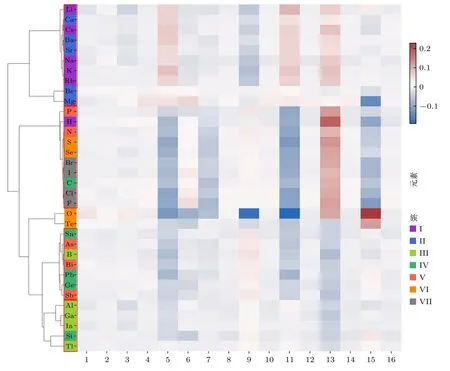
图4 主族元素层次聚类图Fig.4.A hierarchical clustering diagram of the main family elements.
另外,为了更好地理解原子特征向量在高维空间中的内涵,利用主成分分析的方法,将16 维原子特征向量降维到4 个主成分.分别做出第一主成分(PCA1)、第二主成分(PCA2)和第三主成分(PCA3)、第四主成分(PCA4)的散点图,如图5 和图6 所示.在图5 中,可以看到,原子特征向量在第一个主成分上的投影基本上可以反映原子最外层价电子数目,最外层价电子数目越多,第一主成分值越大.在图6 中,第三、四主成分的散点图可以将主族元素按同族元素的相似的化学性质进行聚类.

图5 PCA1 和PCA2 散点 图Fig.5.PCA1 and PCA2 scatter maps.

图6 PCA3 和PCA4 散点图Fig.6.PCA3 and PCA4 scatter maps.
综上所述,利用深度学习在大量材料中提取的原子特征向量可以表示元素的信息,在下一节中将验证本文提取到的原子特征向量在材料信息学的下游任务中有较好的应用.
4 用于预测任务
为了验证基于深度学习得到的原子特征向量(Atom_DL)的有效性,利用得到的原子特征向量对MXY Janus 单分子层材料的带隙进行预测.MX2型过渡金属硫化物(其中M 代表过渡金属,X是硫族元素)在大量文献中得到讨论[19].最近实验[20]表明,可以通过用两个不同的卤族、硫族或磷族元素来合成金属原子M 两侧的X,这形成了一类新二维材料,称为MXY Janus 单分子层.从C2DB数据库[21−22]中得到216 个MXY Janus 单分子层材料数据,用机器学习的方法来预测MXY Janus单分子层的带隙.由于C2DB 数据库中材料带隙值的缺失,最终筛选出93 条MXY Janus 单分子层材料数据进行预测任务.
机器学习模型的输入包括材料的原子特征和材料结构特征.分别将基于Magpie 方法、Atom2Vec方法和深度学习的方法得到的原子特征向量作为材料的原子特征输入模型,其中Magpie 方法①https://github.com/hackingmaterials/matminer/tree/46d6a90664dc9e804e81c2c22cbee9e7221e8315/matminer/utils/data_files/magpie_elementdata和Atom2Vec 方法②https://github.com/idocx/Atom2Vec的数据分别可以在两个开源项目中获得.Magpie 方法利用已知的原子物理化学性质,可以简单高效地构造每个材料的特征向量;但是使用该方法进行预测任务时,往往难以统一特征向量不同分量的量纲.Atom2Vec 方法是一种分布式表示方法,这种方法得到的原子特征向量是连续的、低维的,并且特征向量各分量量纲统一;但是这种方法使用前需要先在大型数据集上预训练.对于材料的结构特征,考虑到MXY Janus 单分子层具体的结构性质,选择材料3 个原子的归一化相对位置和晶胞面积作为材料的结构特征输入模型.在输入模型时,将材料的原子特征和材料结构特征拼接起来作为输入特征,来预测材料的带隙值.
利用3 种机器学习方法(随机森林、核岭回归、支持向量回归)对MXY Janus 单分子层的带隙性质进行建模和预测.随机森林回归模型[5]通过随机抽取样本和特征,以并行的方式获得多棵相互不关联的决策树的预测结果,对所有决策树的预测结果取平均值,作为随机森林回归模型的预测结果.
核岭回归[23]就是基于核函数并且包括l2范数的线性回归.对于线性回归模型,可以使用最小二乘法计算回归模型的参数,但是当样本数据中存在多重共线性的问题时,参数数值会变得非常大.在最小二乘法回归模型的基础上添加参数的l2范数,即为岭回归的目标函数:
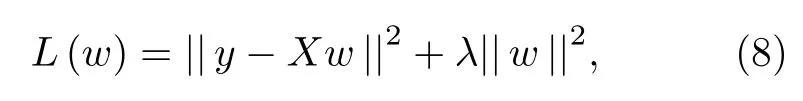
其中λ大于0.为了最小化目标函数,对(8)式右边关于参数w求导,并且令导数为0,即可得到参数w的最优解为

这里Id为单位矩阵.对于非线性数据,通过非线性映射函数Φ将低维空间的数据映射到高维空间,也就是用Φ(X) 代替X,使数据线性可分.在岭回归中加入核函数K,即为核岭回归.重复岭回归求解过程,可以得到核岭回归参数的最优解为
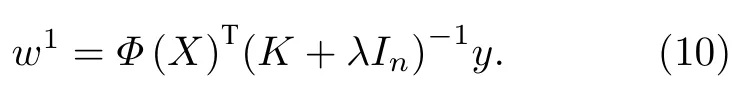
支持向量回归[24]求解一个线性超平面,使得特征空间中的所有样本点到该超平面的几何间隔最大.本质上是求解一个有约束的优化问题,其目标函数为

其中w是回归模型的参数,ε是容忍偏差,y是样本真实值.支持向量回归和线性回归的一个重要区别就是,支持向量回归存在一个容忍偏差,只有当回归模型预测值和真实值的差大于容忍偏差,才计算损失.在求解优化问题时,通过拉格朗日乘子法求解.对于非线性数据,支持向量回归和核岭回归类似,通过引入核函数将数据从低维空间映射到高维空间,使之可以求解.
以上3 种机器学习模型各有特点,随机森林回归由于随机性,可以有效降低模型的方差,具有较好的泛化能力和抗过拟合能力;核岭回归通过增加正则化项,提升了训练的稳定性,具有可解释性、泛化能力强等优点,并且适用于小样本数据回归;支持向量回归作为一种监督学习算法,具有很好的泛化能力,并且对异常值具有鲁棒性.
为了得到稳定的结果,利用5 折交叉验证对模型进行检验.5 折交叉验证将数据集平均划分成5 份,依次用其中的一份作为测试集,其他数据作为训练集来得到误差.最后,计算5 个误差的平均值作为模型最终的误差.在不同的机器学习方法和不同的输入原子特征向量组成的模型中,使用参数搜索的方式得到最优的模型.相同的机器学习方法的参数在同一个参数空间中进行搜索,所有的机器学习算法模型都是使用开源库Scikit-learn[25]实现的.随机森林模型的参数如表1 所示,核岭回归的参数如表2 所示,支持向量回归模型参数如表3所示.

表1 随机森林模型参数Table 1.The random forest model parameter.

表2 核岭回归模型参数Table 2.Kernel ridge regression model parameter.
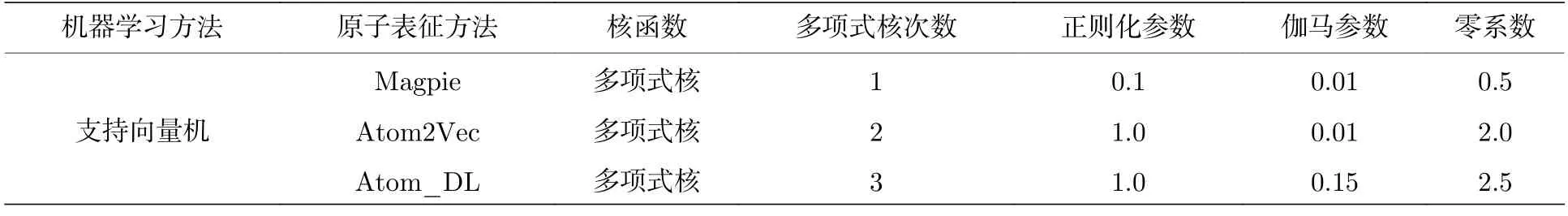
表3 支持向量机回归模型参数Table 3.Support vector regression model parameter.
各模型的预测结果的平均绝对误差如图7 所示,对于3 种机器学习方法,基于深度学习得到的原子特征向量作为输入特征时的平均绝对误差要低于已有的两种原子特征向量表示方法.此外,当Magpie 和Atom2Vec 方法得到的原子特征向量输入机器学习模型中时,核岭回归模型预测的精度是最差的;而对于本文提出的原子特征向量方法,核岭回归模型预测的精度比其他两种方法高.由于核岭回归更适用于处理特征相关性高的数据集,而Magpie 得到的原子特征向量分量量纲不统一,特征相关性自然很低,所以平均绝对误差比较高;而本文提出的原子特征向量量纲统一,各特征之间有一定的相关性,所以平均绝对误差较低,这也说明本文得到的原子特征向量是低维、密集的分布式特征向量.
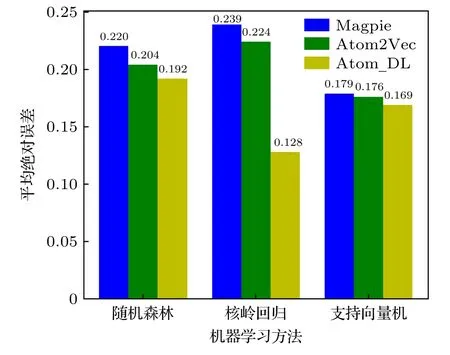
图7 对MXY Janus 单分子层材料的带隙预测平均绝对误差Fig.7.MAE of band gap prediction for MXY Janus monolayer materials.
表4 列出了测试集中的24 个样本的带隙计算值和带隙预测值,其中带隙计算值是通过基于密度泛函理论的第一性原理计算得到的,在密度泛函理论中使用PBE 交换关联泛函进行计算,自旋轨道耦合通过非自洽对角化包含在Kohn-Sham 特征态的全基中[22].从表4 可以看出,3 种机器学习模型均对基于深度学习的原子特征向量具有良好的预测效果.这也验证本文得到的原子特征向量的在机器学习方法中的有效性,这样得到的原子特征向量可以应用到其他材料性质预测的任务中.
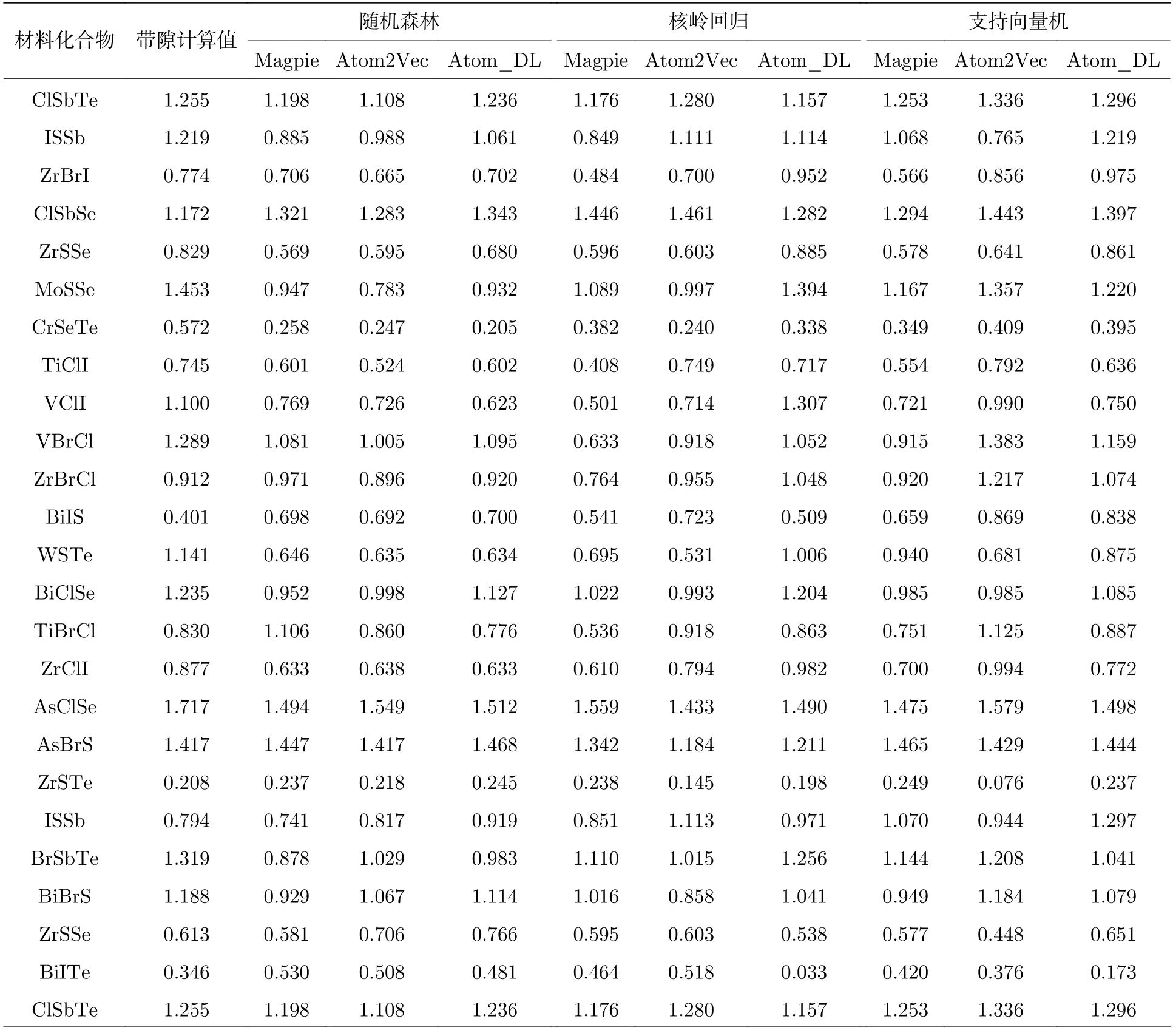
表4 测试集材料预测值和计算值对比Table 4.Comparison of material predictive and experimental values in the test.
5 结论
本研究基于性质相似的原子可以和同样的原子形成结构和性质相似的化合物的观点,利用深度学习的方法从大量材料化学式数据中提取主族元素和大多数副族元素的原子特征.使用随机森林、核岭回归和支持向量回归3 种机器学习方法对Janus 结构过渡金属硫化物MXY Janus 单分子层材料的带隙性质进行预测.在材料特征表示上,使用了材料结构特征和原子特征.材料结构特征使用组成化合物各原子的归一化相对位置和晶胞面积,原子特征分别使用Magpie,Atom2Vec 和Atom_DL.为了得到回归效果更好的模型,对每一种机器学习模型定义一个相同的参数搜索空间,使用Scikit-learn 库中的参数网格搜索函数在参数搜索空间中进行搜索,得到机器学习模型的最佳参数,使用该参数对测试集上的材料数据进行预测,计算测试集的平均绝对误差.从结果上来看,基于深度学习提取到的原子特征在机器学习模型中表现出更好的性能.
随着机器学习的不断发展,机器学习模型在材料信息学中的应用越来越广泛.而利用机器学习模型的第一步就是特征工程,所以本研究结果可以应用到其他材料任务中去.在材料的特征表示上,材料的结构特征对性能的影响也不容忽视,也将关注提取不同类型材料的材料结构特征.
猜你喜欢
九江职业技术学院学报(2022年1期)2022-12-02 09:46:54
新高考·高一数学(2022年3期)2022-04-28 07:02:46
保定学院学报(2022年2期)2022-04-07 02:26:50
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
中学生数理化(高中版.高考数学)(2021年1期)2021-03-19 08:28:36
许昌学院学报(2018年4期)2018-05-02 12:27:37
中华建设(2017年1期)2017-06-07 02:56:14
高中生学习·高三版(2016年9期)2016-05-14 09:12:05
