
中国成人肥厚型心肌病诊断与治疗指南2023
2023-02-13 01:46国家心血管病中心心肌病专科联盟中国医疗保健国际交流促进会心血管病精准医学分会中国成人肥厚型心肌病诊断与治疗指南2023专家组
中国循环杂志 2023年1期
国家心血管病中心心肌病专科联盟、中国医疗保健国际交流促进会心血管病精准医学分会“中国成人肥厚型心肌病诊断与治疗指南2023”专家组
肥厚型心肌病(hypertrophic cardiomyopathy,HCM)可能是第一个基于病因与发病机制进行精准治疗的心血管病,越来越受到心血管专科医师的重视。2017 年《中国成人肥厚型心肌病诊断与治疗指南》(2017 指南)发表后[1],有力地推动了我国HCM 临床诊疗工作的规范化开展。近年来,随着HCM 在病因、发病机制、诊疗手段、危险分层、康复管理等方面的知识与技术创新,迫切需要把这些科学技术与临床研究的成就,介绍给临床医师以及需要的患者。国家心血管病中心心肌病专科联盟、中国医疗保健国际交流促进会心血管病精准医学分会组织国内本领域长期从事HCM 以及相关疾病工作的专家,更新了《中国成人肥厚型心肌病诊断与治疗指南2023》。和2017 指南比较,本指南更新要点如下:
更新要点
(1)更新了HCM 的定义,“拟表型”疾病不再包括在HCM 之中,减少临床混淆
(2)完善了HCM 的临床分型,有利于指导临床对HCM 的认知和诊疗
(3)增加了对HCM 病程特点的论述,有利于对疾病危险分层和预后评估的理解
(4)在诊断方法部分,结合近年的进展,更详尽、全面、深入地介绍了超声心动图、心脏磁共振成像(cardiac magnetic resonance imaging,CMR)、放射性核素显像、病理检查等方法,有助于HCM 的精准诊断和精准干预
(5)作为HCM 的重要诊断方法,基因诊断部分,针对HCM 定义的更新,简化了致病基因列表,便于掌握;加入了本领域新的循证证据,并给出了具体推荐;同时列出了基因诊断流程,便于临床医师执行
(6)增加了HCM 诊治流程的总体框架,便于临床医师实施
(7)将心脏性猝死(sudden cardiac death,SCD)危险分层与防治单列为一节,以方便临床应用和评估
(8)治疗方面,结合近年来药物、介入、外科手术治疗的新理念、新方法进展,做了相关介绍和推荐
(9)随着对HCM 治疗和康复理念认识水平的提高,将生活方式管理和随访单列一章,有助于临床医师和患者把握
(10)根据对HCM 患者个体化管理的要求,新设多学科合作章节,有利于对HCM 患者更加精准的分级诊疗和防控等策略的实施
本指南的相关建议仅供专业人员临床决策时参考,在临床的具体诊疗工作中还需要遵循“个体化”的原则。
为便于读者了解某一项诊断或治疗的价值,本指南对推荐类别/证据水平的表述沿用国际通用的方式。
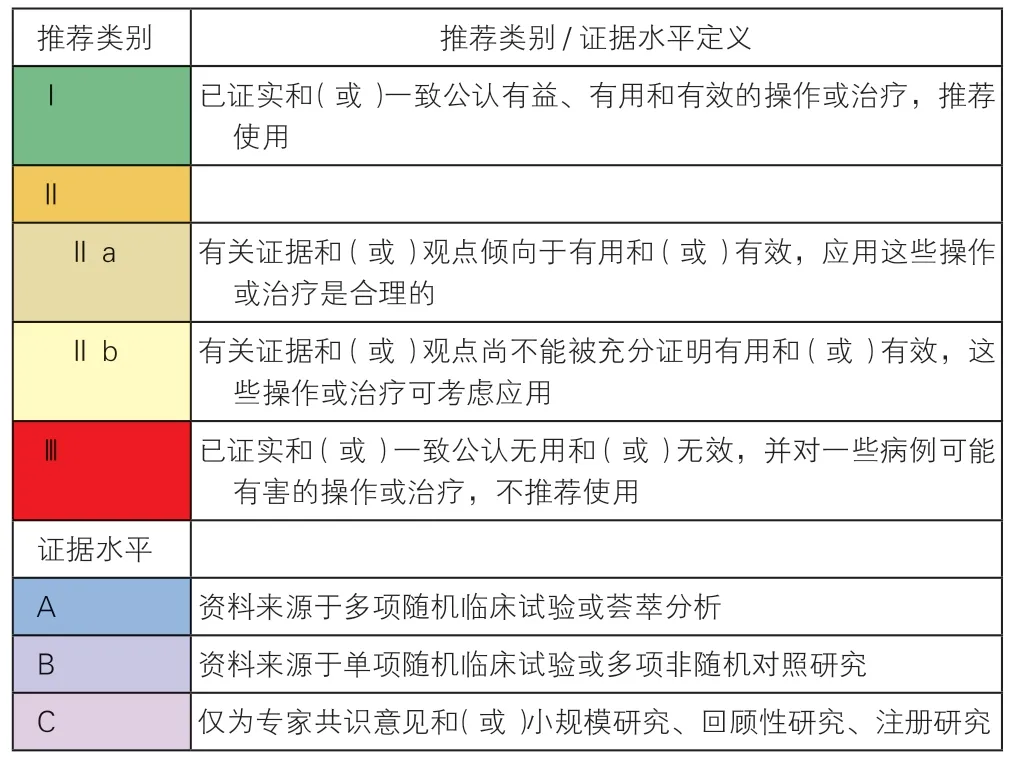
推荐类别/证据水平的分级及定义
1 定义和分型
HCM 主要是由于编码肌小节相关蛋白基因致病性变异导致的、或病因不明的以心肌肥厚为特征的心肌病,左心室壁受累常见,需排除其他的心血管疾病或全身性、代谢性疾病引起的心室壁增厚。超声心动图或者磁共振检查左心室舒张末期任意部位室壁厚度≥15 mm 可确诊,致病基因检测阳性者或者遗传受累家系成员检查发现左心室壁厚度≥13 mm也可确诊[2]。
HCM 可根据血流动力学、遗传学特点或者肥厚累及的部位进行分型。
1.1 根据血流动力学特点
这种分型有利于指导患者治疗方案的选择,是目前临床最常用的分型方法。
(1)梗阻性HCM:异常肥厚心肌突入左心室腔,造成血流通道阻塞,并在其上下方产生左心室流出道压力阶差(left ventricular outflow tract gradient,LVOTG)。根据LVOTG 的变化情况分为静息梗阻性和隐匿梗阻性,前者指静息时LVOTG 峰值≥30 mmHg(1 mmHg=0.133 kPa),后者指静息时LVOTG 峰值<30 mmHg 而激发后LVOTG 峰值≥30 mmHg[3]。心肌肥厚累及右心室时,静息时右心室流出道压力阶差峰值≥16 mmHg 诊断为右心室流出道梗阻[4]。
(2)非梗阻性HCM:静息时或激发后LVOTG峰值均<30 mmHg。
1.2 根据遗传学特点
分为家族性HCM 和散发性HCM。家族性HCM是指除先证者外,三代直系亲属中有一个或以上成员被确诊为HCM,或存在与先证者相同的基因变异,伴或不伴有心电图及超声心动图异常。否则为散发性HCM。
1.3 根据心肌肥厚部位
(1)心室间隔肥厚:临床最常见,主要累及室间隔基底部。部分累及室间隔中部,表现为左心室中部乳头肌水平的室间隔肥厚。
(2)心尖部肥厚:主要累及左心室乳头肌水平以下心尖部,通常不伴LVOTG 升高。
(3)左心室壁弥漫性肥厚:少数患者表现为左心室壁弥漫性增厚。
(4)双心室壁肥厚:除左心室壁肥厚外,还有右心室壁肥厚(右心室游离壁厚度>5 mm)[4]。
(5)孤立性乳头肌肥厚:主要特点是乳头肌肥厚,其余左心室节段不受影响[5]。
2 流行病学
20 世纪90 年代国外基于超声心动图筛查的研究显示HCM 在成人中患病率为1∶500[6]。21 世纪初中国在8 000 余名普通人群中超声心动图筛查HCM患病率约为80/100 000[7]。但早期筛查手段的限制导致HCM 患病率很可能被低估。随着临床和分子遗传学研究的不断深入,尤其是家族谱系筛查的推广以及更敏感的心脏影像学诊断的实施,HCM 的患病率据估计至少为1/200[8]。中国医学科学院阜外医院单中心研究发现,HCM 从2016 年起成为该院门诊和住院患者最常见的心肌病类型,占心肌病门诊及住院患者的45%,并且呈持续上升态势[9]。来自普通人群筛查性研究获得的患病率与临床诊断HCM 患病率之间的差异,说明相当一部分无症状HCM 患者可能未被确诊。
3 病因和发病机制
HCM 的主要病因是编码肌小节蛋白或肌小节相关结构蛋白的基因变异,主要表现为常染色体显性遗传,约60%的HCM 存在致病性或可能致病性基因变异[10],仍有大约40%的HCM 未找到明确致病基因。
肌小节是心肌纤维的基本单位,其成分包括粗肌丝、细肌丝、Z 线、M 线等。编码粗肌丝相关的β-肌球蛋白重链基因(β-myosin heavy chain,MYH7)和心脏型肌球蛋白结合蛋白C 基因(myosin binding protein C,MYBPC3)是HCM 最主要的致病基因,约各占HCM 的15%~30%。目前已证实的基因变异超过2 000 余种[8]。
基因变异可以通过改变氨基酸序列,即显性负效应(毒肽效应),产生具有生物学功能缺陷的蛋白;也可以通过降低编码蛋白的表达水平(单倍型剂量不足),使正常蛋白合成不足,最终造成肌小节或肌小节相关蛋白结构或功能异常,如Ca2+敏感性增加、ATP 酶活性异常、肌球-肌动蛋白相互作用或肌小节装配发生改变等,使得心肌收缩异常、舒张功能受损、能量消耗增加,进而引起心肌压力感受及应答通路异常,诱发心肌细胞的组织学和形态学变化,导致心肌细胞肥大、排列紊乱、间质纤维化、心肌重塑等[10-11]。
虽然HCM 被传统观点认为是单基因疾病,但是,HCM 的最终临床表型是基因型、修饰因子以及环境条件等多种因素共同作用的结果。相同的基因变异因个体的基因表现度和外显率不同,以及遗传背景、表观修饰、生活方式或其他暴露因素的差异可呈现不同的临床表型[10-11]。部分HCM 病因及发病机制尚不明确,对其相关的探索仍在不断进行之中。
4 诊断
4.1 临床表现
4.1.1 症状
不同专业学员各项考核成绩比较见表1。儿内科和儿外科学员各项技能成绩比较显示,不同科室的学院骨髓穿刺术成绩对比,差异具有统计学意义[91.0(86.2,92.8) vs.87.9(19.6,89.9),(P=0.008)],其余项目考核成绩比较,差异无统计学意义(P>0.05)。
HCM 临床症状变异性大,有些患者可长期无症状,而有些患者首发症状就是猝死。儿童或青少年时期确诊的HCM 患者症状更多,预后可能更差。
(1)呼吸困难:是最常见的症状,多为劳力性呼吸困难[12]。主要是由左心室舒张期充盈压升高,以及升高的左心室压力传回肺循环所致。左心室充盈压升高是心室壁肥厚导致舒张顺应性受损和心内膜下缺血共同作用的结果。
(2)胸痛:大约40% 的HCM 患者有胸痛不适的症状[13],在不合并冠状动脉粥样硬化的HCM 患者中也很常见。这是由于心肌肥厚,心室舒张受损和心肌耗氧量显著增加,导致心肌缺血所致。
(3)头晕:常劳累时加重,可能因劳力后LVOTG 增大,或活动后汗液蒸发,血容量下降导致。头晕也可能是由于快速站立或排便时的 Valsalva 动作引起。某些药物,如利尿剂、硝酸甘油和血管扩张性药物会增加LVOTG,加重左心室流出道梗阻。头晕也可能继发于非持续性心律失常相关的低血压和脑灌注减少。
(4)心悸:多与心功能减退或心律失常有关。HCM 患者常合并心律失常,例如房性和室性期前收缩、窦性停搏、心房颤动(房颤)、心房扑动(房扑)、室上性心动过速和室性心动过速(室速)等。
(5)晕厥:15%~25% 的HCM 患者至少发生过一次晕厥,另有20% 的患者有先兆晕厥,一般见于活动时[1]。儿童和青年人常常在运动时出现,由于运动时心输出量不足或合并心律失常而引起。非持续性房性或室性快速性心律失常是HCM 患者常见的晕厥原因,而部分HCM 患者存在窦房结和房室结功能异常,会导致严重的心动过缓,也是引起晕厥的重要原因。伴有晕厥的患者猝死风险增加,需要积极检查和治疗。
(6)SCD:HCM 是青少年和运动员发生SCD 最常见的病因[14],尤其与过度劳累有关。在超过 80%的病例中,导致猝死的心律失常是心室颤动(室颤)。
4.1.2 体征
HCM 体格检查所见与患者疾病状态有关。典型体征与左心室流出道梗阻有关,无或梗阻较轻的患者可无明显的阳性体征。
触诊:心前区心尖搏动常常横向移位,心尖搏动通常范围大,异常有力。
听诊:第一心音正常。第二心音通常是正常分裂的,但在流出道压力阶差大的患者中,可闻及矛盾分裂的第二心音。S3 奔马律在儿童中很常见,但在成年人听到S3 奔马律时,提示失代偿性心力衰竭(心衰)。
梗阻性HCM 患者胸骨左缘第3~4 肋间可闻及较粗糙的喷射性收缩期杂音,杂音通常是收缩期渐强-渐弱杂音,不向颈部传导,增加心肌收缩力(如运动、室性期前收缩后)或减轻心脏前负荷的措施(如站立位、Valsalva 动作、含服硝酸甘油等)可使杂音增强;相反,减弱心肌收缩力或增加心脏前负荷措施(如β 受体阻滞剂或蹲位、抬腿等)可使杂音减弱。收缩期二尖瓣前叶前向运动(systolic anterior motion,SAM)征和存在明显LVOTG 增高的患者可闻及二尖瓣关闭不全的心尖部全收缩期杂音,向腋窝传导。
4.1.3 病程特点
HCM 的临床病程具有多样性,近一半的患者初诊时无明显的临床症状[15],只是被偶然发现。而在另一些患者,HCM 则沿着特定的疾病进程发展,穿插着改变疾病病程的临床事件,并影响治疗策略。在三级医疗中心就诊的HCM 患者年死亡率为2%~4%[16];来自选择性较低的社区HCM 患者年死亡率为1.5%~2.0%[17];而在规范管理策略干预下,有可能将年死亡率降至0.5%[18]。由于多数HCM 患者及基因携带者病程很长,在全生命周期中会合并多个合并症,这些合并症会明显影响患者的死亡率和死亡原因。如合并冠心病的HCM 约占10%~53%[19],由于冠心病与HCM 临床表现常常类似,有时会忽视其诊治,其自然病程会明显改变。
SCD、心衰和血栓栓塞是HCM 死亡的三大主要原因。SCD 是HCM 灾难性的临床结局,其危险性随年龄增长可能逐渐下降,但不会消失[16]。SCD 的早期识别和危险分层是HCM 首要的临床问题,本指南有专门章节进行论述。
HCM 患者血栓栓塞的最常见原因是房颤,患病率20%左右[20],相关年死亡率约0.7%。研究显示,未经抗凝治疗或依从性差的患者,血栓栓塞事件的发生率比接受规律抗凝治疗的患者高出7 倍以上[21]。
心衰是HCM 广泛存在的并发症,与HCM 患者死亡风险增加独立相关[22]。HCM 的心衰以射血分数保留的心衰(heart failure with preserved ejection fraction,HFpEF)为主,主要由心室舒张功能异常引起。心室舒张过程包括主动舒张和被动充盈,主动舒张是指心肌内在的特性,被动充盈与心肌僵硬度(或顺应性)有关。HCM 患者既有心肌主动舒张障碍,也有被动充盈障碍。研究显示43.5%的HCM患者可进展为HFpEF[22]。HCM 患者由于心室壁增厚,心腔相对较小,左心室射血分数(left ventricular ejection fraction,LVEF)为50%时实际搏出量可能不足以满足循环需求,因此,当LVEF 接近50%,就应警惕收缩功能不全。
部分患者可进展至HCM 终末期,表现为严重左心室收缩功能障碍,主要诊断标准是超声心动图检查LVEF ≤50%,伴或者不伴左心室扩大。HCM终末期患者往往反复出现心衰症状,如果患者达到NYHA 心功能分级Ⅲ~Ⅳ级,预后比HFpEF 差[23],甚至比扩张型心肌病患者更差[24]。
临床上有部分HCM 患者呈限制型表现,超声心动图诊断标准为:(1)双心房明显扩大,超声多普勒提示E/A 比值≥2 和减速时间≤150 ms,房颤患者满足后者即可;(2)无或轻度左心室肥厚;(3)心室腔缩小或正常;(4)LVEF 正常或轻度降低[25]。HCM 人群中限制型表现的发生率为1.5%~5.9%[25-26]。与典型HCM 比较,该类患者临床表现重,心衰和猝死率高,生存率和特发性限制型心肌病患者接近[25]。
HCM 患者肺动脉高压发生率在18%~50%之间,年发病率为1%。合并肺动脉高压的HCM 患者生活质量和活动耐量更差,病程长和左心室收缩功能减退是肺动脉高压发生的独立预测因素[27]。运动相关肺动脉高压(运动过程中肺动脉收缩压≥60 mmHg)近年来受到越来越多的关注,有文献报道HCM 患者运动相关肺动脉高压发生率为38%,出现运动相关肺动脉高压的患者左心室舒张功能更差、静息肺动脉收缩压更高、N 末端B 型利钠肽原(N-terminal prohormone of B-type natriuretic peptide,NT-proBNP)水平更高[28]。肺动脉高压、运动相关肺动脉高压均为HCM 患者预后不良的独立危险因素[29],应当早期识别并及时干预。
肾功能不全是HCM 患者常见的并发症。有研究提示HCM 患者中超过半数估算肾小球滤过率(estimated glomerular filtration rate,eGFR)<90 ml/(min·1.73 m2),约15% 患者eGFR <60 ml/(min·1.73 m2)。HCM 合并肾功能不全在女性、高龄及左心室收缩功能异常的患者中更加常见[30]。这一类患者除风险增加外,其临床诊断需要更加谨慎,注意与多器官、多系统受累的疾病相鉴别。
4.2 诊断方法
4.2.1 实验室检查
(1)生物学标志物:①B 型利钠肽或NTproBNP:可以协助诊断HCM 是否合并心衰以及对疾病进展与预后进行评估[31](Ⅰ,B)。②心肌肌钙蛋白:HCM 患者心电图可表现有ST-T 改变及异常Q波(Ⅰ,C)。超半数HCM 患者可存在血清心肌肌钙蛋白不同水平升高,且与心功能相关[32]。其升高可能与HCM 患者存在心肌细胞隐匿性损伤有关。③反映炎症、纤维化、细胞死亡的标志物,如肿瘤坏死因子α、基质金属蛋白酶等,有利于HCM 疾病的危险分层与预后评价。多项指标的组合检测可能是未来发展的方向。
(2)血常规和血生化检查:血常规、血生化指标、铁代谢指标等应作为HCM 患者初诊常规检查项目(Ⅰ,C),用于评估患者基础状况及是否合并其他疾病。如涉及鉴别诊断,则可检测与疾病明确相关的特殊指标,如免疫固定电泳、KAPPA 轻链、LAMDA 轻链用以鉴别心脏淀粉样变等(Ⅱa,C)。
4.2.2 心电信息检查
(1)常规心电图:虽然当今多模态心脏影像技术发展迅速,但心电图仍是HCM 患者不可或缺的初步评估手段,并可能是疾病早期唯一异常表现。常规心电图具有灵敏度高、简便易行等特点,可提供各种心律失常、心房/心室肥大以及心肌缺血等信息。HCM 患者出现常规心电图异常的比例可高达90%以上。作为初筛和随访的主要手段之一,推荐对所有疑诊和确诊的HCM 患者进行常规心电图检查(Ⅰ,B)。
HCM 心电图异常通常有以下表现:①心房异常:由于舒张功能减低、二尖瓣反流等导致心房扩大,心电图可有相应表现。②病理性Q 波:在HCM患者中,病理性Q 波通常认为存在透壁心肌纤维化,发生率18%~53%。③左心室高电压:HCM 患者心电图出现左心室高电压的比例约为43%~60%,同时单独出现左心室高电压而无其他心电图异常的发生率很低(1%~2%),这可为与其他疾病的鉴别诊断提供依据。④复极异常:ST-T 改变在HCM 中的发生率高,部分表型如心尖HCM 可有典型的巨大倒置T 波等特征性改变。⑤QT 间期延长:1/8 的HCM 患者可出现QT 间期延长,并可能与埋藏式心脏复律除颤器(implantable cardioverter defibrillator,ICD)放电密切相关[33]。
对HCM 鉴别诊断的研究发现,如果同一导联Q 波和正向T 波同时出现,或者前侧壁导联出现巨大倒置T 波,则倾向诊断HCM;如果出现PR 间期缩短和(或)心室预激波,则更支持糖原贮积病或法布雷病;如果左心室壁增厚伴肢体导联低电压,则倾向诊断心脏淀粉样变[34]。
(2)动态心电图监测:动态心电图能够连续记录受检者是否存在快速或者缓慢型心律失常、传导阻滞、窦性停搏及其持续时间、严重程度等。由于HCM 易合并心律失常,推荐所有HCM 患者行24~48 小时动态心电图监测,以评估心律失常、SCD的风险[35](Ⅰ,B)。
(3)有创电生理检查:心脏电生理检查可记录和标测心内心电图,通过各种特定的电脉冲刺激来评估心律失常的起源部位并行相应的治疗。一般不推荐HCM 患者应用有创电生理检查作为疾病危险分层和评判预后的手段(Ⅲ,C)。而对于存在预激综合征,或考虑行器械治疗心律失常的HCM 患者,可考虑行电生理检查[36](Ⅱb,C)。由于存在诱发多形性室速的风险,所以术前应充分评估,且操作应在经验丰富的心血管中心进行。
4.2.3 负荷试验
负荷试验可用于评估左心室流出道是否存在隐匿性梗阻,或评估运动或心肺贮备功能,用以指导疾病的远期管理和治疗。对于静息状态没有而运动后有流出道梗阻症状的HCM 患者,推荐使用运动负荷试验以排除隐匿性梗阻(Ⅰ,B);室间隔减容术前评估可考虑在有经验的临床医师指导下行运动负荷试验(Ⅱb,C)。运动负荷检查过程中应密切关注患者症状和体征,并配备相应的急救人员和设施。若运动负荷试验无法进行,可采取药物激发试验替代[37-38](Ⅰ,B)。
心肺运动试验可用于HCM 与生理性心肌肥厚(如运动员)的鉴别诊断(Ⅰ,B)。对HCM 危险分层、心脏康复训练指导和预后判断具有一定价值,也是心脏移植受者筛选的重要评估手段[37-38](Ⅱa,B)。心肺运动试验禁用于梗阻性、心律失常高风险或血流动力学不稳定的HCM 患者(Ⅲ,C)。
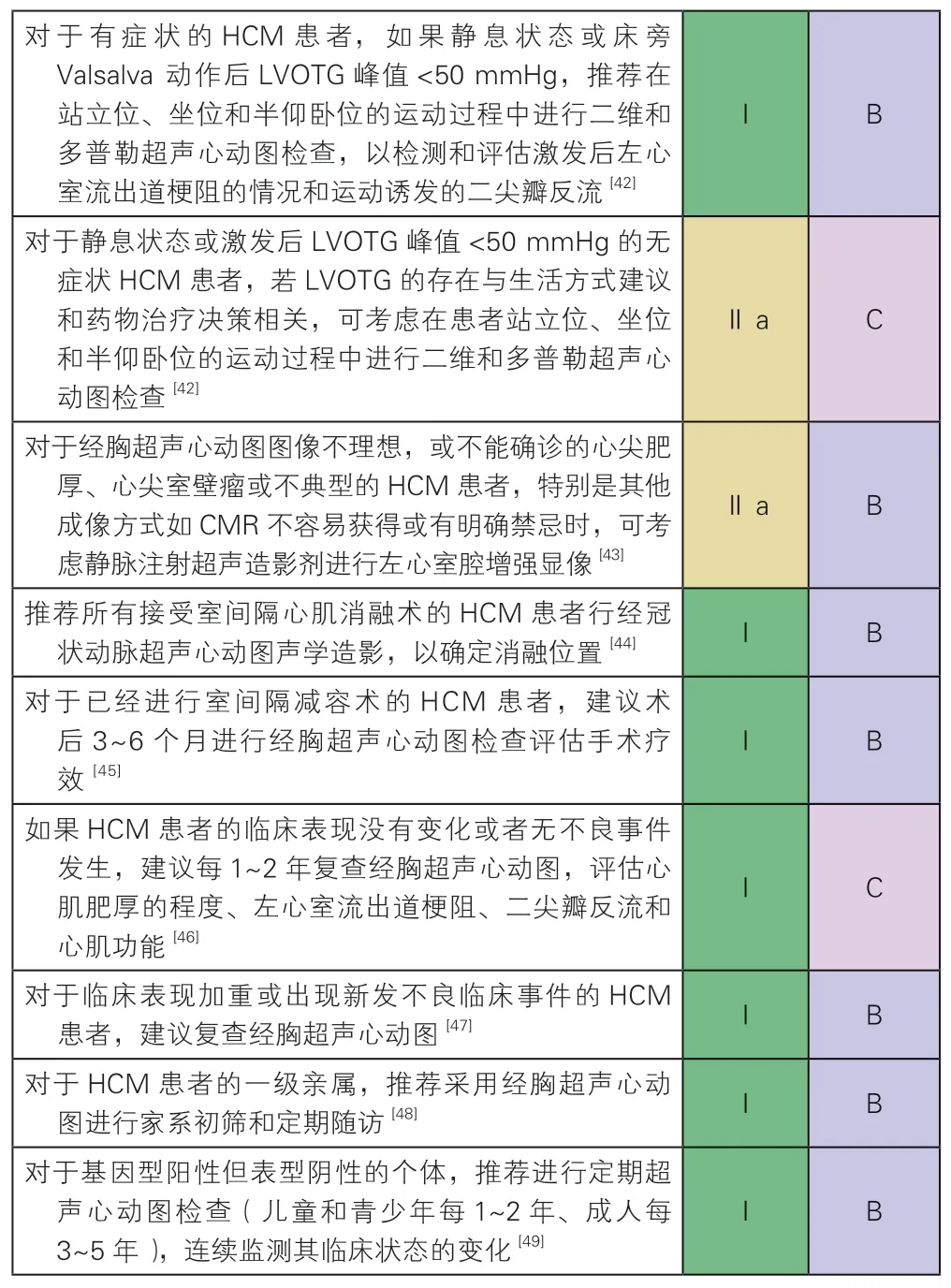
4.2.4 超声心动图
超声心动图是HCM 诊断首选、准确且经济的方法,所有HCM 患者均应行经胸超声心动图检查,必要时还需进行心脏超声造影和经食道超声心动图检查(Ⅰ,B)。
(1)评估左心室壁厚度:HCM 心室壁肥厚可发生在任何部位,对于已确诊或可疑HCM 的患者,需要在舒张末期左心室短轴二尖瓣、乳头肌和心尖切面进行连续心室壁厚度的测量。
(2)评估左心室流出道梗阻:HCM 患者LVOTG 是动态变化的,受各种改变心肌收缩力和心脏负荷因素的影响。因此,对静息状态无左心室流出道梗阻的患者,需要在激发状态下进行评估。当检测到左心室腔内压力阶差时,需系统扫查确定有无左心室中部梗阻、二尖瓣器异常以及主动脉瓣下隔膜。
(3)评估SAM 征和二尖瓣反流:SAM 征导致瓣叶对合不良,继发收缩中晚期为著及偏后外侧的二尖瓣反流;测量反流速度和时间可帮助与左心室流出道湍流鉴别。
(4)评估左心室舒张功能:HCM 患者多伴有明显的左心室舒张功能障碍,评估其左心室充盈压有助于评估症状,判断疾病分期。一般采用多普勒超声心动图对HCM 患者左心室舒张功能进行综合评价。
(5)评估左心室收缩功能:有研究显示,HCM患者LVEF 正常时或携带致病基因变异的家属在心室壁尚未增厚前,就出现了左心室纵向收缩速度和应变减低[39]。采用组织多普勒心肌显像和斑点追踪成像技术评估心室各节段纵向收缩速度和形变参数(应变和应变率),能够更准确地评估HCM 患者的心室收缩功能。
(6)HCM 鉴别诊断:某些特定的超声心动图特征有助于疾病的诊断。向心性肥厚常见于法布雷病;双心室壁肥厚和双心室流出道梗阻多见于Noonan 综合征;心肌呈斑点或颗粒状回声、少量心包积液、房间隔增厚、主动脉瓣结节状增厚、LVEF轻度减低伴限制性充盈多提示淀粉样变。

HCM 经胸超声心动图检查推荐建议
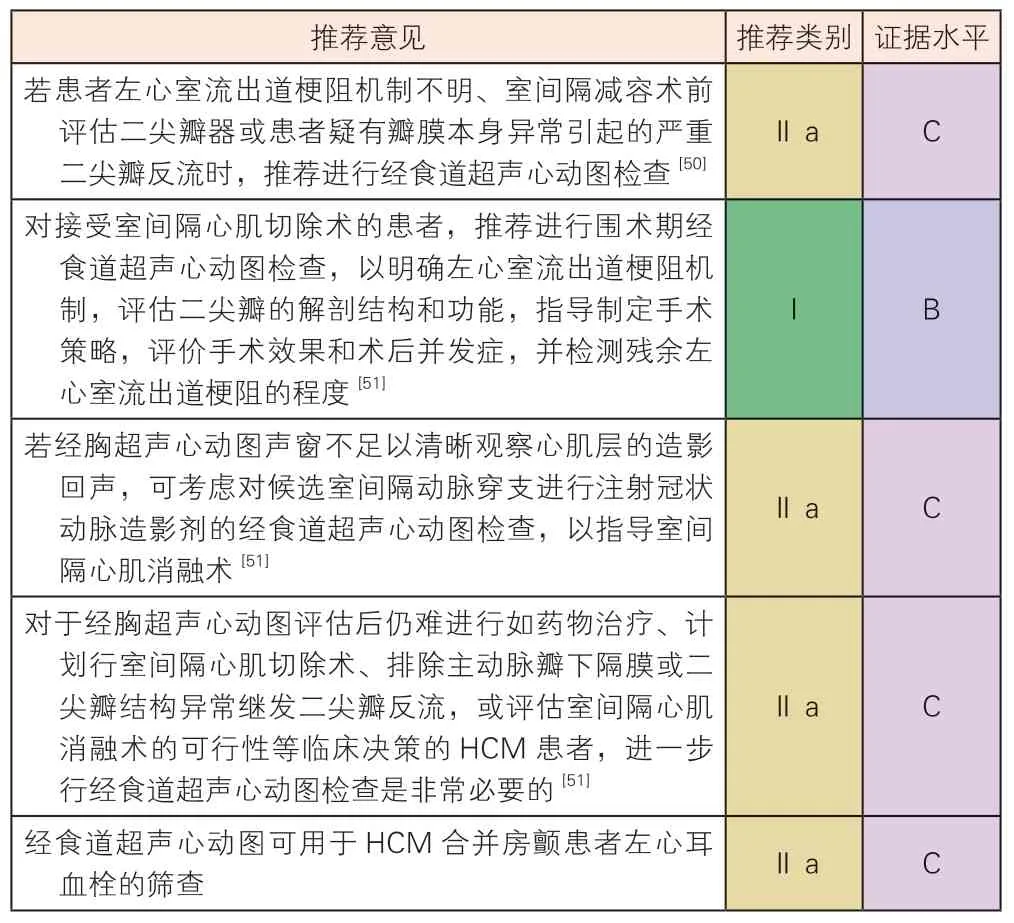
HCM 经食道超声心动图检查推荐建议
4.2.5 心脏磁共振成像
对比超声心动图,CMR 除了能够准确显示心脏结构与功能变化外,还可以结合钆对比剂延迟强化(late gadolinium contrast enhancement,LGE)在体识别心肌纤维化[52]。CMR 能清晰显示左心室壁肥厚的节段性改变,如心尖HCM 伴有的心尖心腔裂隙状、囊状突出或伴室壁瘤形成等。心尖HCM 合并室壁瘤并不少见,需要与左心室中段HCM(乳头肌水平环性增厚)相鉴别[53]。对称性HCM 表现为左心室壁弥漫性增厚,需排除任何引起左心室后负荷增加的心肌疾患,并且还需与系统性或代谢性疾病心肌受累相鉴别。
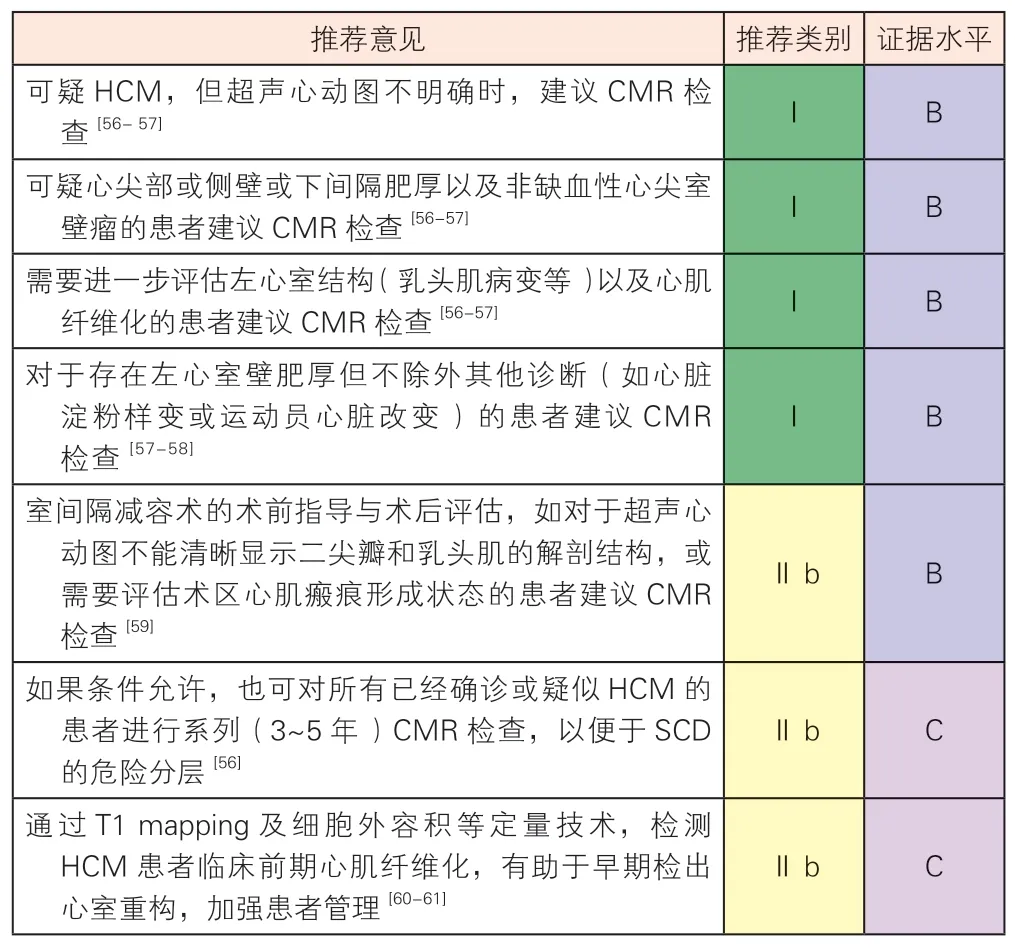
CMR 在HCM 诊断中的推荐建议
LGE 是目前临床在体评估心肌纤维化最有效的方法,约有65% 的HCM 患者会出现LGE,多表现为肥厚心肌内局灶性或斑片状强化,其中以室间隔与右心室游离壁交界处局灶状强化最典型[54]。室间隔非对称性肥厚伴右心室插入部强化以及继发性左心房扩大是HCM 最常见征象。LGE 在疾病预后判断和危险分层中发挥重要作用,出现广泛LGE(LGE定量≥左心室质量的15%或目测LGE 分布广泛)提示SCD 风险增加[55]。
4.2.6 放射性核素显像
放射性核素显像在HCM诊断中可用于以下情况:

放射性核素显像在HCM 诊断中的推荐建议
(1)心肌缺血和冠状动脉微循环功能障碍的鉴别:心肌缺血是HCM 的病理机制之一,对患者预后有重要影响。利用核素心肌灌注显像可以无创检测HCM 是否存在明显的心肌缺血,对于有胸部不适但冠心病可能性低的HCM 患者,利用核素心肌灌注显像诊断是否合并冠心病具有较好的阴性预测值[62]。另一方面,即使证实无心外膜下冠状动脉病变,HCM 患者仍有可能存在冠状动脉微循环障碍及冠状动脉血流储备降低。目前的心肌灌注显像技术,无论是应用正电子发射断层显像还是单光子发射断层显像,均能实现心肌血流的定量分析,获得负荷和静息状态下的心肌绝对血流量,从而计算冠状动脉血流储备[63],是评估冠状动脉微循环障碍重要的影像学方法[64]。此外,应用心肌灌注显像可以检测心尖部的心肌梗死,预测心尖部室壁瘤形成。
(2)心脏淀粉样变的鉴别:放射性核素骨显像可用于鉴别有心肌肥厚表现的转甲状腺素蛋白淀粉样变,其显像剂结构多为99锝m(99Tcm)标记的磷酸盐衍生物,如:99Tcm-标记焦磷酸盐(99Tcmpyrophosphate,99Tcm-PYP)、99Tcm-标记双羧基双膦酸盐和99Tcm-标记羟基亚甲基二磷酸盐等,目前国内主要应用99Tcm-PYP。99Tcm-PYP 可与心脏淀粉样纤维中的钙与磷酸盐特异性结合,如果心肌放射性摄取为2 级(与骨摄取相当)或3 级(高于骨摄取),则为阳性,提示淀粉样变累及心肌[65]。虽然另一类免疫球蛋白轻链心脏淀粉样变也会有部分患者心肌有较低的99Tcm-PYP 摄取,一般为1 级(低于骨摄取)或2 级,但如果结合血清/尿液中单克隆免疫球蛋白阴性,则99Tcm-PYP 显像诊断淀粉样变的特异性和阳性预测值接近100%,几乎可以替代心肌活检[66]。对于免疫球蛋白轻链心脏淀粉样变,18氟(18F)-哌啶醇、18F-氟比他班和11C 匹兹堡复合物B 等特异性显像剂目前在研究中也显示出了较好的显像效果[67],未来有希望应用于心脏淀粉样变的精准分型。
4.2.7 计算机断层血管成像/造影
对于有心绞痛或存在心肌缺血证据的HCM 患者,建议行冠状动脉计算机断层血管成像/冠状动脉造影用以排除是否存在缺血性心脏疾病。对于需要行减容术的HCM 患者,推荐冠状动脉造影检查用以评估冠状动脉的解剖和狭窄情况[68-69](I,B)。
4.2.8 左心室造影
左心室造影检查不仅可通过造影显示心脏和血管的形态结构,还可测量心腔内的压力。存在左心室流出道梗阻时,左心室造影可见心室腔与流出道之间存在收缩期压力阶差。左心室造影可以帮助判断特殊类型HCM,如心尖肥厚患者心腔呈典型的“黑桃A”形态。存在以下情况的HCM 患者推荐行此检查(Ⅰ,B):(1)非侵入性影像学检查无法明确是否存在左心室流出道梗阻;(2)HCM减容治疗(包括室间隔心肌消融术与外科切除术)的术前评估[70-71]。
4.2.9 病理检查
(1)心内膜心肌活检:心内膜心肌活检(心肌活检)病理检查有助于诊断或鉴别诊断疑似的代谢性或系统性疾病心肌受累。适应证包括:①HCM 合并心衰[72](Ⅱb,C);②不明原因的室壁增厚,无创检查及心肌组织以外的其它组织活检仍不能除外代谢性或系统性疾病心肌受累,如转甲状腺素蛋白和轻链型淀粉样变性、庞贝病、法布雷病[2,73](Ⅱb,C)。禁忌证:①出血性疾病、严重血小板减少症及正在接受抗凝治疗者;②急性心肌梗死、心室内附壁血栓形成者禁忌行该心室活检;③左心室室壁瘤形成者禁忌左心室活检;④近期有急性感染者;⑤不能很好配合的患者。
(2)HCM 的病理特征:HCM 以左心室和(或)右心室壁肥厚、尤其是室间隔的非对称性肥厚为特征。左心室腔常减小,室间隔基底部的肥厚可导致左心室流出道梗阻,也可见左心室中段肥厚和心尖肥厚。可合并二尖瓣器异常,例如乳头肌直接插入前瓣瓣叶、乳头肌融入心壁、副乳头肌等。镜下可见心肌细胞肥大、排列紊乱伴间质纤维化,心壁内小动脉常见结构不良改变和狭窄。其中排列紊乱是HCM 的重要组织学特征,但是单独出现不能作为诊断或排除HCM 的依据,因为其分布的不均一性和正常心脏局部或部分继发左心室壁肥厚病例也可见到排列紊乱。间质纤维化的程度可随病程演变而明显增加,呈现围细胞的、小片状的或广泛的纤维化等多种形态,室间隔受累常重于左心室游离壁。部分病例的替代性纤维化或纤维瘢痕较显著,可掩盖心肌细胞排列紊乱,干扰心肌活检病理诊断[74]。
(3)与HCM 具有鉴别诊断意义的其他疾病心肌受累的病理特征:心脏淀粉样变常在心肌间质、心肌细胞周围和血管壁沉积,刚果红染色阳性,偏振光显微镜检查可见特征性的“双折光”。心肌活检是淀粉样变的确诊手段和分型的基础(免疫组织化学染色,显微切割与质谱分析)。法布雷病心肌组织学可见显著的空泡变性,电镜检查可见大量电子致密的同心性层状小体即髓鞘样小体。庞贝病心肌可出现不同程度的空泡变性,过碘酸雪夫染色阳性,电镜下可见大量膜包绕的糖原小体。Danon 病心肌细胞内可见嗜碱性颗粒,可以被非特异性酯酶和乙酰胆碱酯酶染色,过碘酸雪夫染色阳性,电镜检查可见自噬体堆积,2 型溶酶体相关膜蛋白免疫组织化学染色可完全缺失。线粒体疾病可有酶组织化学改变,电镜下可见线粒体增生和巨大线粒体或线粒体包涵体[75-76]。
4.3 基因诊断
4.3.1 概述
基因变异是绝大多数HCM 患者的根本病因,约60% 的HCM 患者可以找到明确的致病基因变异[2,73,77-78],因此基因检测对指导HCM 诊治有重要临床意义。
目前认为HCM 主要是由罕见变异通过常染色体显性遗传模式导致的疾病,偶有隐性遗传模式报道。已经报道与HCM 相关的基因众多(表1),但证据充分的明确致病基因主要为编码肌小节蛋白的基因,包括MYH7、MYBPC3、TNNT2、TNNI3、MYL2、MYL3、TPM1和ACTC1,这些基因的变异可以解释超过90% 的遗传阳性HCM,因此,HCM也被称为“肌小节疾病”。其中以MYH7和MYBPC3基因变异所占比例尤多,约占所有肌小节变异患者的80%~90%。除了MYBPC3基因外,导致HCM 的肌小节变异主要以错义突变为主,而MYBPC3基因的致病变异超过半数为插入/缺失或剪接位点变异,导致蛋白截短表达。也有研究显示除了经典的罕见变异通过单基因遗传模式致病外,常见变异的复杂遗传模式可能也是HCM 的患病原因,尤其是对于上述已知致病基因阴性的患者,应予以关注[79-80]。
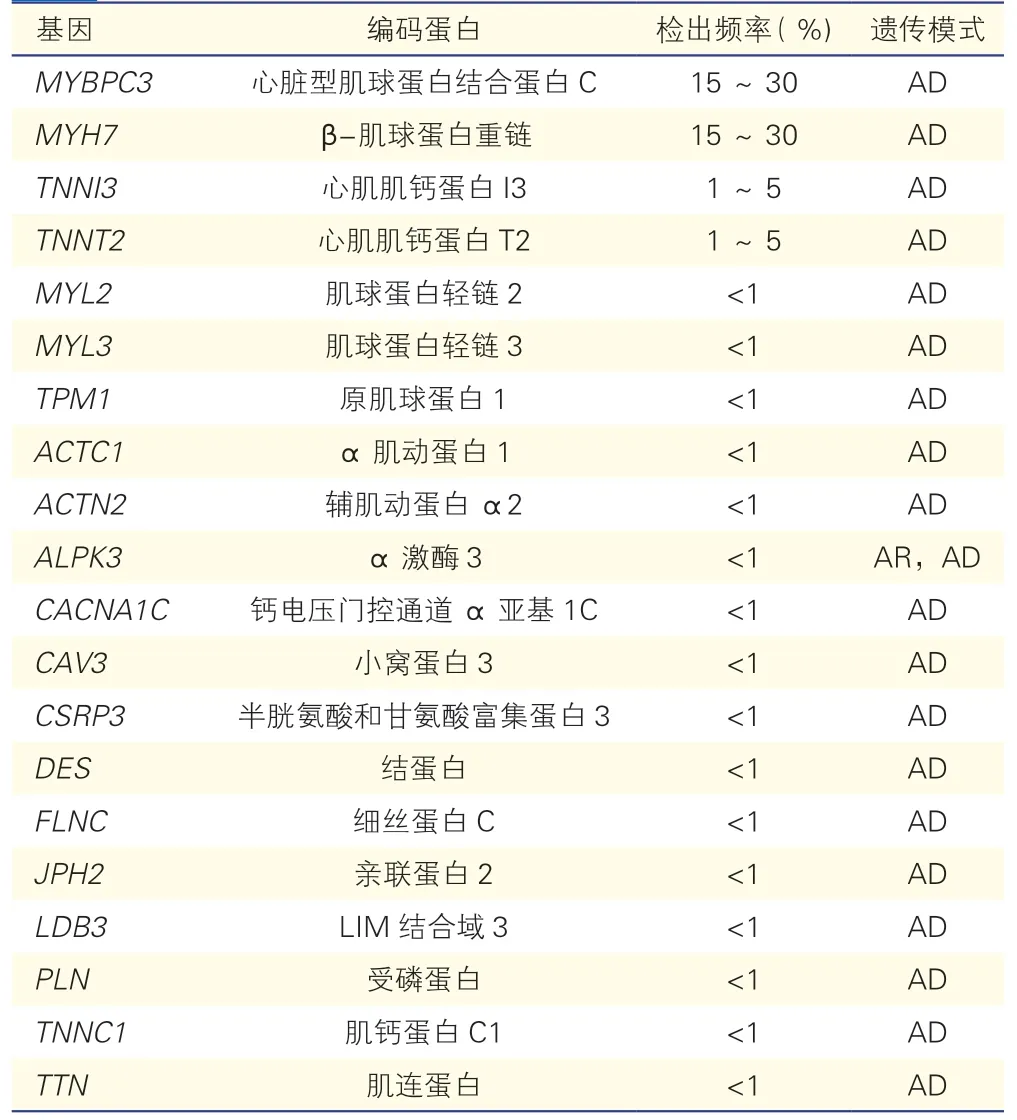
表1 肥厚型心肌病致病基因
另外,部分代谢性疾病或系统性疾病,包括淀粉样变、糖原贮积病、溶酶体贮积病、线粒体疾病、神经肌肉疾病、血色病、畸形综合征等,会单独导致或伴有左心室壁肥厚[1],也应在HCM 基因检测时纳入,有利于鉴别诊断。
HCM 致病基因的外显率(即致病变异携带者最终发生HCM 的比率)为40%~100%,发病年龄异质性也较大[81],对基因诊断结果解释应谨慎。应在系统收集分析HCM 患者家系(绘制包含三代亲属的家系图)基因型和临床表型信息后,进行规范的遗传咨询[2]。HCM 基因诊断流程见图1。
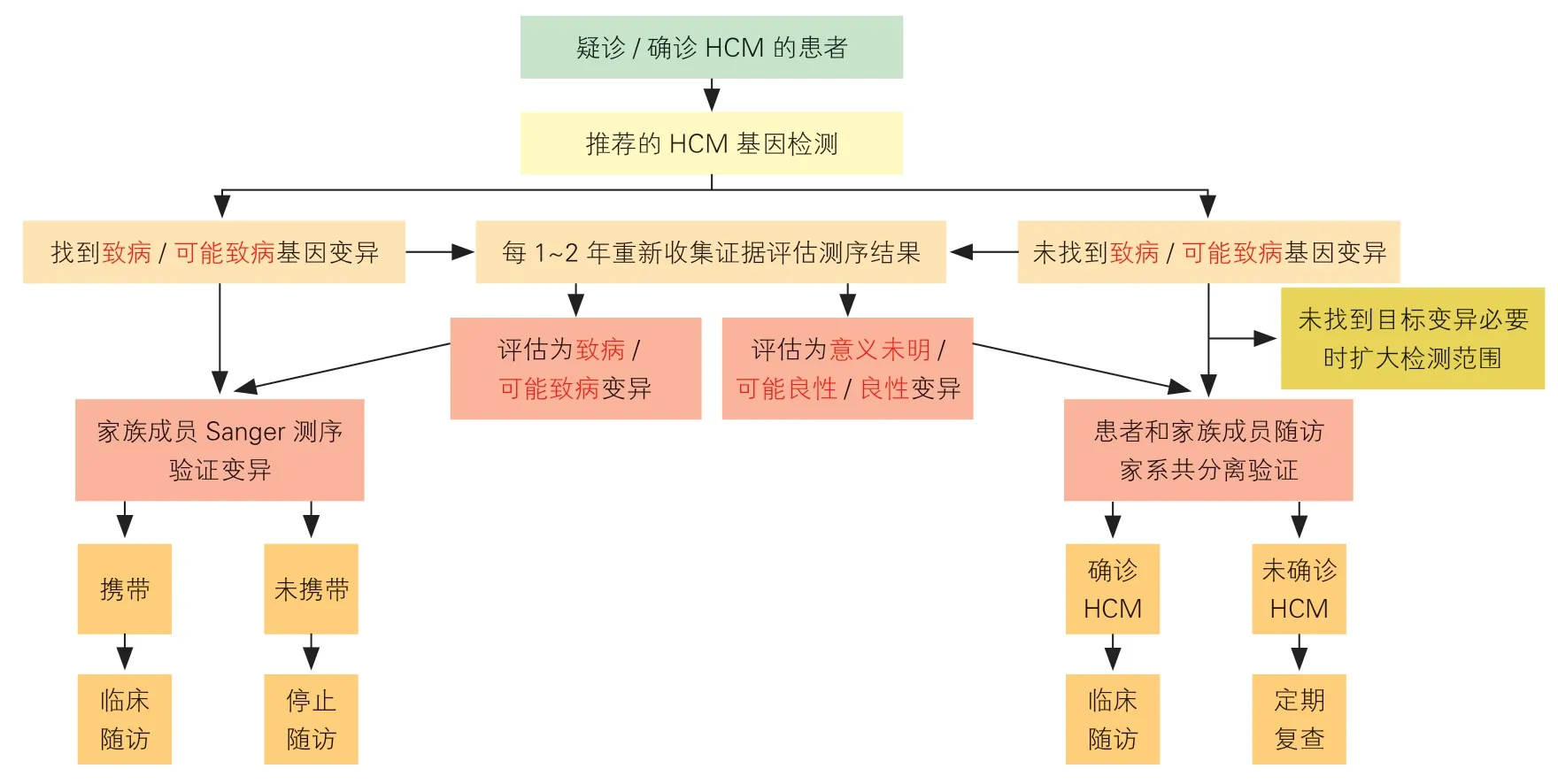
图1 HCM 基因诊断流程
4.3.2 先证者的遗传咨询
家族中第一个确诊为HCM 的患者称为“先证者”。无论是否行进一步临床诊疗或基因筛查,推荐所有HCM 患者进行遗传咨询[2](Ⅰ,B)。遗传咨询应由受过遗传专业训练的遗传专业人员或具有遗传知识的心血管医师进行,分别从医学、遗传学、心理学、伦理学、法律等多个角度与患者进行沟通和提供咨询意见,使其充分了解并学会如何应对HCM 的遗传问题。

HCM 先证者基因诊断推荐建议
通过遗传咨询,也能更好地收集其他家庭成员的相关信息,进而完善家系图谱,并为下一步的病因学检测提供证据和线索。
4.3.3 先证者基因诊断
4.3.4 先证者亲属的基因筛查
应确定 HCM 患者直系亲属(至少三代)是否临床受累或者遗传受累。应全面收集直系亲属的临床病历资料,重点关注有猝死、心律失常、“先天性”心脏病的亲属,即使未见明显心肌肥厚。与先证者充分沟通HCM 的详细病情、遗传风险、对生活与工作的影响、对后代的影响等有助于先证者与亲属沟通。
对于检测到明确致病基因变异的家庭,如果先证者筛查出明确的致病变异,其直系亲属无论是否具有临床表现,均推荐Sanger 法一代测序,验证此致病变异[2,73](Ⅰ,C)。未携带致病变异的亲属一般无需临床随访;携带致病变异而临床尚未有表现的家系成员,则需仔细地进行临床评估(包括超声心动图、心电图、肌钙蛋白、NT-proBNP 等),并长期随访。约7%的患者存在复合或多基因变异,临床表型比较重。如相关亲属的HCM 临床表现与先证者有明显差异,则建议行多基因深度靶向测序[90-91](Ⅰ,C)。
对未检测到明确致病基因变异的家庭,如果先证者检测结果为阴性,或检测到ACMG 分级临床意义未明的变异,其一级亲属应行详细的临床检查(包括超声心动图、心电图、肌钙蛋白、NT-proBNP 等)。由于存在外显延迟,亲属需定期临床复查[2](Ⅰ,C)。对于轻度心肌肥厚达不到诊断标准的年轻亲属,可以每隔6~12 个月进行1 次临床检查(Ⅱa,C),如数次检查病情无进展,可延长复查时间。亲属主诉有相关症状时应重新进行临床评估[2]。
4.4 鉴别诊断
临床上引起心肌肥厚的原因多样,对于出现心肌肥厚的代谢性或者系统性疾病的患者,在治疗上与HCM 存在本质区别。因此,需予以系统的鉴别诊断,优化患者的管理(I,C)。
4.4.1 高血压
高血压导致的心肌肥厚,患者多有长期的高血压病史,心肌肥厚通常呈对称性,超声显示肥厚心肌为均匀的低回声,一般室壁厚度≤15 mm,室间隔与左心室游离壁的厚度比多小于1.3。经严格血压控制 6~12 个月后,左心室壁肥厚程度一般可减轻或者消退[92]。临床上高血压及HCM 两种情况重叠的患者并不少见,对于诊断不明确及治疗效果不佳的患者筛查HCM 致病基因有助于鉴别诊断。
4.4.2 主动脉瓣狭窄和先天性主动脉瓣下隔膜
主动脉瓣狭窄中20%~30% 合并不对称性心肌肥厚,心肌肥厚的程度通常相对较轻(室壁厚度≤15 mm),主动脉瓣狭窄程度常为中度以上,而HCM 患者一般无明显的主动脉瓣病变。超声心动图可明确病变。先天性主动脉瓣下隔膜也常合并心肌肥厚,但心肌肥厚主要表现为对称性,超声心动图和CMR 检查见瓣下隔膜可确诊。
4.4.3 冠心病
HCM 患者出现不典型心绞痛和心电图ST-T 改变、病理性Q 波及广泛对称的倒置T 波,在缺乏其他相关检查结果的情况下易误诊为冠心病,二者需进行鉴别诊断。冠心病患者年龄多在40 岁以上,有高血压、高脂血症等相关危险因素,可并发左心室壁或室间隔肥厚和左心室舒张功能受损。但冠心病患者R 波电压一般不高,超声心动图通常不出现明显的非对称性左心室壁肥厚、左心室流出道梗阻或SAM征。冠状动脉造影及HCM基因检测可协助诊断。
4.4.4 强化运动
运动可诱发生理性心肌肥厚,心电图上可有高电压、早期复极、非特异性ST-T 改变等,但一般无 HCM 家族史、心肺运动功能较好,超声心动图可显示左心室壁轻度均匀增厚(非对称性心肌肥厚或者心尖肥厚罕见),可合并左心室腔内径增大,但通常不合并左心房增大和严重的左心室舒张功能异常,CMR 显示无明显心肌纤维化,终止体能训练可使肥厚程度减轻[93]。
4.4.5 内分泌异常
肢端肥大症时由于生长激素和胰岛素样生长因子-1 分泌过多,会刺激肌小节蛋白合成从而导致心肌肥厚,通常为左心室壁均匀肥厚伴有心腔扩张,且较早出现严重的收缩功能障碍,检测生长激素及生长因子水平可以鉴别[94]。过度分泌肾上腺髓质激素的疾病(如嗜铬细胞瘤),由于激素对心肌细胞的刺激及继发血压升高,也常导致均匀性心肌肥厚伴心腔扩大和室壁运动异常(弥漫或者心尖部的显著运动减低),部分患者心血管表现可在原发病治疗后得到显著改善[95]。糖尿病导致的心肌病是糖尿病患者的一种特殊心脏表现,早期可表现为左心室壁肥厚和舒张功能障碍,并可发展为收缩功能降低和心衰[96]。有文献报道1 型糖尿病母亲分娩的婴儿中有50%、2 型糖尿病中有25% 出现左心室壁肥厚,这些婴儿在脱离患糖尿病的母体后左心室壁肥厚可以消退[97]。在部分甲状腺功能减退相关的心肌病患者中,可表现有室间隔的不对称性肥厚,并可在早期规范的甲状腺激素替代治疗后恢复[98]。
4.4.6 药物因素
长期使用一些药物,包括羟氯喹、他克莫司和促蛋白合成类固醇等,可以导致左心室壁肥厚,但室壁厚度很少会超过15 mm,大部分患者左心室壁肥厚可在停药后逆转[99-100]。羟氯喹是一种抗风湿药物,可能通过抑制溶酶体水解酶而导致心肌病变,主要表现为左心室扩大、室壁肥厚伴收缩功能减退[101]。他克莫司是一种抗移植排斥的免疫抑制药物,文献报道儿童移植患者应用过程中可引发左心室壁肥厚甚至流出道梗阻,停用该药后左心室壁肥厚可以逆转[99-100]。相关发病机制已被归因于细胞内钙处理的改变,部分是通过持续激活钙敏感信号转导途径,逐渐导致心肌肥大和心肌病表型。促蛋白合成类固醇是人工合成的类似雄性激素的药物,它能促进肌肉增长,增强体力,常为一些运动员所服用,使用期间左心室壁轻度增厚,停止服用后室壁厚度恢复正常[100]。此外,抗癌治疗(如蒽环类药物、抗微管类药物、免疫检查点抑制剂、放疗等)可以通过多种机制诱发不同程度的心肌肥厚,可能与药物/射线直接的心脏毒性、代谢重塑/失调以及对心血管和免疫稳态的干扰有关,在有相关治疗的患者中应注意鉴别[102]。
4.4.7 淀粉样变
淀粉样变是由遗传、变性和感染等因素引起蛋白前体形成不可溶性淀粉样纤维并沉积于器官或组织细胞外,导致其结构和功能障碍的一组疾病,其中心脏是淀粉样变常累及的器官,表现为心肌肥厚和舒张功能受损。蛋白前体有不同来源,与分型和预后密切相关[103]。
累及心脏的淀粉样变最常见的有三种类型:(1)因异常浆细胞分泌的单克隆免疫球蛋白轻链沉积引起的轻链型淀粉样变心肌病;(2)基因变异导致肝脏合成分泌的参与转运甲状腺素和维生素A 的转甲状腺素蛋白异常,引起其发生解离、形成淀粉样纤维并沉积的突变型转甲状腺素蛋白淀粉样变心肌病,为常染色体显性遗传;(3)在无基因突变的情况下转甲状腺素蛋白发生解离沉积,导致野生型转甲状腺素蛋白淀粉样变心肌病,多见于70 岁以上男性[104]。
与HCM 不同,淀粉样变导致的左心室壁肥厚通常为对称性,大多数不伴有左心室流出道梗阻,心电图表现为低电压或者正常电压。除心室肌外,房间隔和瓣膜也可以由于淀粉样物质沉积发生增厚。CMR 成像显示LGE 多发生在心内膜下,可以延展至附近心肌,而HCM 时LGE 则多见于明显增厚的室壁处的中层[105]。99Tcm标记的磷酸盐衍生物对于诊断转甲状腺素蛋白淀粉样变心肌病有较好的特异度和灵敏度[106]。另外淀粉样变常有心脏外表现,如周围神经病变、腹泻或者假性肠梗阻、尿蛋白或肾功能不全、玻璃体混浊和双侧腕管综合征等。组织病理能够发现组织间质内无细胞结构的均匀物质沉积。刚果红染色后在偏振光显微镜下观察呈特征性苹果绿色双折光表现。基因检测有助于突变型转甲状腺素蛋白淀粉样变的诊断[106]。
老年人新发的左心室壁肥厚、特别是合并有心电图低电压表现、房室阻滞、主动脉瓣中重度狭窄和心脏外器官受累等情况时,有11%~16% 是由转甲状腺素蛋白淀粉样变所致[106]。
4.4.8 法布雷病
法布雷病是一种X 连锁遗传的溶酶体贮积病,编码α-半乳糖苷酶A 的GLA基因变异引起该酶活性降低或缺失,以致其降解底物——神经鞘脂类化合物及衍生物在心脏等全身多个器官组织细胞中贮积,引起相应多脏器病变,如外周神经疼痛、少汗、皮肤血管角化瘤、蛋白尿、肾功能不全、眼部及心脏病变等。经典型多于儿童期发病,迟发型多为成年后发病,往往男性(半合子)临床表现及分型重于女性(杂合子)。35 岁以上表现为HCM 的患者中0.5%~1.0% 为该病。心脏受累多表现为向心性心肌肥厚,由于神经鞘脂类物质主要沉积在内膜下而肌层受累较轻,超声心动图可见内膜和外膜回声强而中间肌层回声弱的“双边”表现[107]。心电图常表现为传导系统受累,短PR 间期不伴预激综合征,QRS 时限延长,右束支阻滞,此外aVL 导联R 波电压≥1.1 mV 及下壁导联ST 段压低对该病诊断有提示作用[108]。CMR 成像初始T1 值较HCM 有显著降低,T2 值增高,LGE 通常出现在左心室下侧壁基底部,在心肌内正中分布,心内膜和心外膜下常不受累[109]。心肌活检可见心肌细胞肥大,胞浆内空泡变,过碘酸雪夫染色阳性,电镜下胞浆内可见典型的嗜锇性“髓样小体”。需结合α-半乳糖苷酶A 活性、底物及衍生物水平等多项指标检测以及基因检测而明确诊断。
4.4.9 糖原贮积病
糖原贮积病是一组因基因变异导致一种或多种参与糖原合成/降解的酶活性降低或缺乏的遗传病,其特征是组织中糖原沉积或糖原结构异常。该病的鉴别要点主要是多系统受累的临床表现,严重的左心室壁肥厚,早期进展为扩张相,常伴心室预激和传导异常等心电图表现,心肌活检可见心肌畸形肥大排列,多有心脏增大、猝死家族史。
(1)Danon 病:系编码2 型溶酶体相关膜蛋白的基因变异导致的X 连锁显性遗传病。临床主要表现为骨骼肌病、智力发育迟缓和心肌病变。心肌肥厚的患者中0.7%~2.7%经基因检测诊断为该病[110]。男性较女性发病年龄更早,症状更重。心脏受累在男性主要表现为严重的左心室对称性肥厚,而女性多为非对称性肥厚,室壁厚度常达30 mm 以上。心电图左心室高电压明显,80%以上患者合并预激综合征[111]。CMR 成像LGE 分布广泛、形式多样,但以不累及室间隔中段为特征性表现[112]。心肌或骨骼肌活检可见特征性自噬空泡改变,血清肌酸激酶明显升高提示该病,基因检测有助于诊断。
(2)单磷酸腺苷激活蛋白激酶γ2 亚基编码基因变异心脏综合征:系由该基因变异导致的常染色体显性遗传病。临床主要表现为左心室壁肥厚、预激综合征和逐渐进展的传导系统疾病。大部分患者无心脏外表现,少数可有骨骼肌异常。心肌肥厚患者中经基因检测约0.5%诊断为该病[113]。心脏受累早期可呈局限于左心室侧壁、后壁中部的非对称性肥厚,进展期以室间隔为著,通常不伴左心室流出道梗阻和SAM征;利用超声心动图斑点追踪技术(又称靶心图或牛眼图)可见“条带样”特异性改变[114]。基因检测有助于明确诊断。
4.4.10 线粒体疾病
线粒体疾病是因线粒体DNA 或细胞核DNA 缺陷,引起细胞氧化磷酸化功能失调,导致多系统病变的一种遗传疾病。患者可在任何年龄起病,以对能量代谢需求较高的眼、脑、骨骼肌和心脏受累最为常见。心脏病变见于40%的患者,其中心肌肥厚最常见[115]。早期多表现为左心室壁肥厚,之后逐渐出现心脏扩大、LVEF 降低、传导阻滞或心室预激。除心脏受累外,患者通常伴有其他系统受累,包括神经肌肉病变、内分泌、消化系统或肾脏等。实验室检查血乳酸、丙酮酸最小运动量试验阳性;肌肉及心肌活检是指导诊断的有利证据,心肌活检电镜示细胞内大量巨大的异常线粒体聚集,线粒体嵴增多且排列紊乱,细胞色素氧化酶/琥珀酸脱氢酶免疫组化双重染色可见由于部分肌纤维细胞色素氧化酶缺陷所导致的“马赛克”样变化;基因检测发现核DNA 或线粒体DNA 变异等有助于诊断[116]。
4.4.11 Friedreich 共济失调
Friedreich 共济失调是一种由FXN基因第一内含子(GAA)n 发生异常扩增、基因缺失或点突变所致常染色体隐性遗传病[117]。患者多在青春期前后起病,临床主要表现为进行性步态和肢体共济失调、构音障碍、腱反射消失、病理征阳性和骨骼异常。34%~77%的患者伴有心肌肥厚。主要为左心室向心性肥厚,心电图显示有T 波倒置、电轴左偏和复极异常。疾病晚期可出现左心室增大和收缩功能减低,心衰和心律失常是死亡的主要原因[118]。基因检测有助于诊断。
4.4.12 血色病
血色病是一类由过多铁质沉积在组织器官而引起不同程度的基质细胞破坏、纤维组织增生及功能障碍的疾病[119]。原发性血色病主要由HFE、HFE2、HAMP、TFR2、SCL40A1等基因变异导致先天性铁代谢障碍;而继发性则多见于大量输血、长期内服铁剂、慢性酒癖并摄入含铁饮食所致。患者常有铁沉积的系统性损害,如全身皮肤色素沉着、糖尿病及肝硬化等表现。患者早期即可出现左心室壁增厚和心脏舒张功能异常,心电图可有肢体导联低电压,各种心律失常(房性及室上性心律失常多见,房室阻滞和室性快速型心律失常较少见)[120],晚期常进展至左心室扩张和收缩功能改变[121]。转铁蛋白饱和度(>60%)、肝脏和心脏影像学提示铁负荷过度,肝穿刺/心肌活检和基因检测有助于明确诊断。
4.4.13 畸形综合征
畸形综合征主要是指丝裂原活化蛋白激酶途径基因变异引起的遗传性疾病,每1 000 到2 500 名儿童中就有1 人受到影响。丝裂原活化蛋白激酶信号通路有15 个以上的基因编码,对细胞周期至关重要,在增殖、分化、生长和代谢中具有调节作用,会导致一系列疾病包括Noonan 综合征、豹斑综合征、Costello 综合征、心面皮肤综合征[122]。这些畸形综合征可合并心肌肥厚表现,需要与HCM 进行鉴别,鉴别要点在于是否合并心脏外畸形和异常,基因检测发现丝裂原活化蛋白激酶信号通路相关的基因变异有助于诊断。
4.5 诊断流程
临床诊断HCM 应基于以下因素:HCM 家族史、不明原因的症状(如呼吸困难、胸痛、乏力、心悸、晕厥或先兆晕厥)、收缩期喷射性杂音和心电图异常,有上述一个或多个临床发现时,应进一步行超声心动图和(或)CMR 检查等以确定诊断,需排除其他明确的心原性、系统性或代谢性疾病导致的心肌肥厚[1]。诊断流程如图2。

图2 HCM 诊治流程
成人HCM 的临床诊断标准为二维超声心动图或CMR 测量的左心室舒张末任意节段室壁厚度≥15 mm,且无其他已知的可引起心肌肥厚的病因。当患者室壁厚度为13~14 mm,同时伴有HCM 家族史或有基因检测阳性结果时,也可诊断HCM[2]。左心室壁的任一节段都可能出现心肌肥厚,以前间隔基底段和前壁游离壁最易受累,部分人群肥厚部位非常局限,仅累及左心室1~2 个节段,也有患者出现右心室壁肥厚[56]。
尽管单独依靠症状或者体征无法确诊HCM,查体仍是临床医师疑诊此类疾病的最初检查手段[73]。应对所有疑诊HCM 患者进行系统的问诊、查体并对其3 代以内家族史进行详细问询(I,B)。
5 心脏性猝死危险分层与防治
5.1 危险分层方法与防治
SCD 危险分层和预防是HCM 患者临床管理重要的组成部分。猝死常由于室性心律失常引起,目前安装ICD 是公认的预防HCM 患者SCD 最有效和可靠的方法。既往明确发生过SCD 事件,包括心脏骤停、室颤、持续性室速导致意识丧失或血流动力学紊乱的HCM 患者推荐植入ICD 进行SCD 二级预防。而对于HCM 患者SCD 一级预防,国内外尚未形成普遍共识。目前国际上存在多种HCM患者SCD 危险分层方法,2014 年欧洲心脏病学会(European Society of Cardiology,ESC)HCM 诊断和治疗指南推荐使用肥厚型心肌病心脏性猝死风险预测(HCM Risk-SCD)这一数学模型,个体化评估成人HCM 患者5 年SCD 风险,并据此指导ICD安装(图3)。2022 年ESC 室性心律失常和SCD的管理指南仍然肯定了这个模型的预测意义[123]。该模型包括7 个因素:就诊年龄、最大左心室壁厚度、左心房内径、LVOTG、非持续性室速(non-sustained ventricular tachycardia,NSVT)、近期(6 个月内)发生不明原因的晕厥、SCD 家族史,可以使用网络计算器计算相应评分(https://doc2do.com/hcm/webHCM.html)。该模型的计算方法不适用于职业运动员、有代谢性或者系统性疾病导致心肌肥厚的患者、行心肌切除术或者室间隔心肌消融术后的患者[123]。
2020 年美国心脏协会(American Heart Association,AHA)/美国心脏病学会(American College of Cardiology,ACC)HCM 指南推荐根据患者有无下列危险因素评估SCD 风险,并决定是否安装ICD(图3):(1)SCD 家族史;(2)严重的左心室壁肥厚(≥30 mm);(3)不明原因的晕厥;(4)左心室心尖室壁瘤;(5)LVEF<50%;(6)NSVT;(7)CMR 提示广泛LGE。
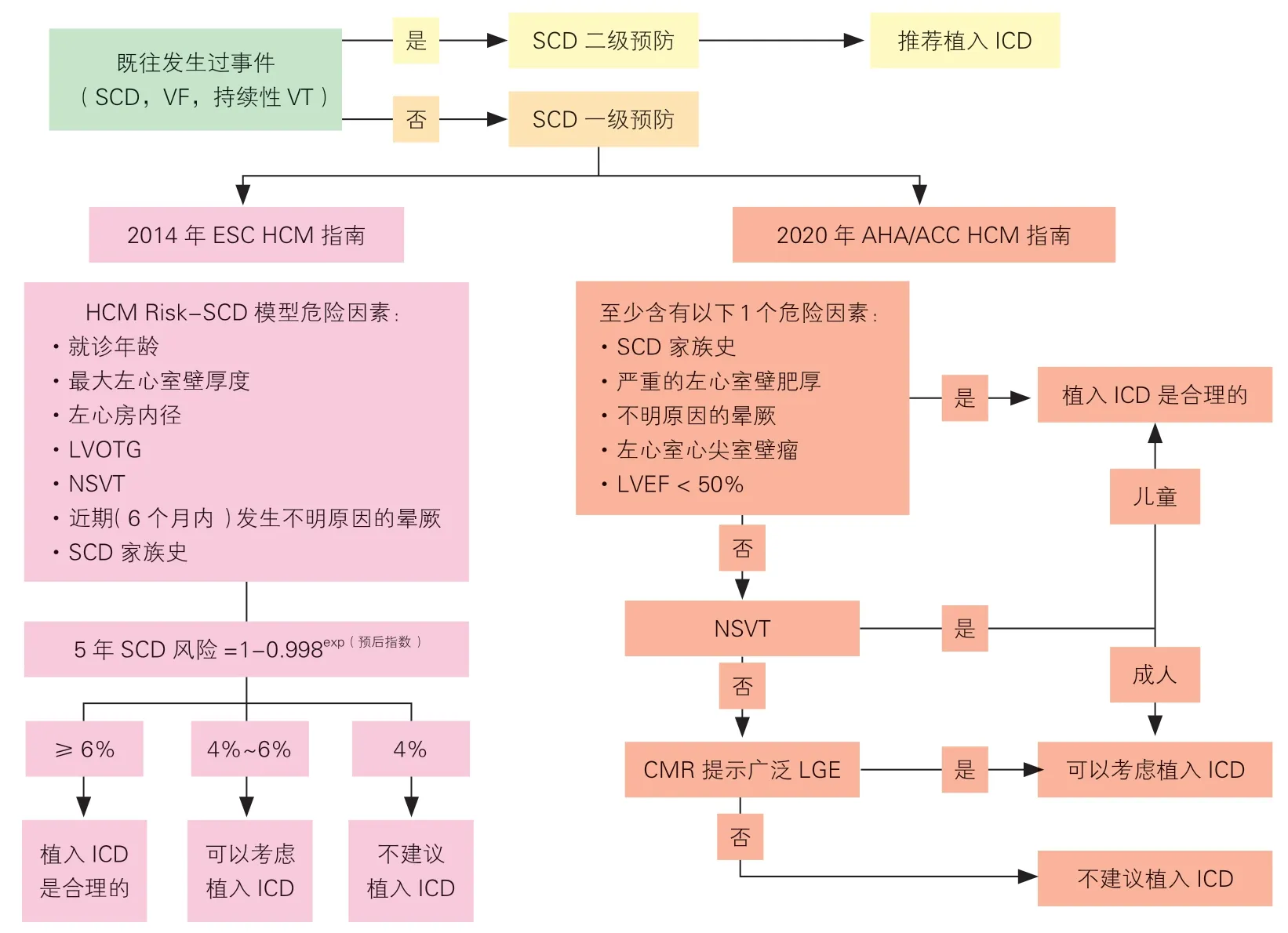
图3 国际上不同的HCM 危险分层模型
国内不同队列研究验证了上述模型的预测能力,研究结果显示在中国HCM 人群中,AHA/ACC的SCD 危险分层方法优于ESC 的HCM-Risk SCD 模型[124-125]。
目前国际上针对儿童HCM 患者的SCD 预测模型有2 个:欧洲学者提出的HCM Risk-Kids 模型个体化评估儿童(1~16 岁)HCM 患者5 年SCD 风险[126]。该模型包括5 个因素:不明原因的晕厥、NSVT、左心房内径Z值、最大左心室壁厚度Z值、LVOTG,可以使用网络计算器计算相应评分(https://hcmriskkids.org)。美国学者提出了PRIMACY 预测模型预测儿童(<18 岁)SCD 风险[127],纳入的变量包括诊断年龄、室间隔厚度Z值、左心室后壁厚度Z值、左心房内径Z值、LVOTG、NSVT 和不明原因的晕厥。目前这两个预测模型在中国儿童HCM 患者中的应用价值尚待进一步研究验证。
5.2 风险评估指标
HCM 患者应该在最初诊断以及每1~2 年进行系统的、全面的非侵入性的SCD 风险评估[128](Ⅰ,B),内容包括:(1)心脏骤停或者持续性室性心律失常的个人史;(2)怀疑心律失常晕厥史;(3)HCM 相关猝死、心脏骤停、持续性室性心律失常的家族史;(4)超声心动图评估最大左心室壁厚度、LVEF、左心房内径、左心室心尖室壁瘤等;(5)动态心电图监测发现的NSVT。经过临床评估后,未定义为高风险患者或者不确定是否安装ICD 的HCM 患者,可以通过CMR 来评估患者最大左心室壁厚度、LVEF、左心室心尖室壁瘤和LGE 心肌纤维化范围[129](Ⅰ,B)。ICD 治疗建议参见室性心律失常部分。
除上述需评估的指标之外,目前也有一些研究指出存在其他危险因素,包括:(1)运动血压反应异常:指从静息到最大运动量血压升高≤20 mmHg或从最大运动量到静息血压降低≤20 mmHg,约20% HCM 患者有运动低血压反应,且40 岁以下患者SCD 的危险增加[130]。(2)发病年龄轻:发病年龄越小SCD 危险越大,尤其是合并NSVT、不明原因的晕厥或严重左心室壁肥厚的患者[128]。(3)左心室流出道严重梗阻:有研究报道LVOTG ≥30 mmHg是SCD 的独立危险因素[131],但尚未达成共识。(4)同时携带多个基因变异:若同一个患者携带≥2 个致病基因变异,无论变异来自同一基因还是不同基因,均可能导致更为严重的临床表型,SCD 风险增加[90-91]。(5)其他:研究发现心肺运动试验中LVEF下降、心率反应异常、峰值摄氧量下降的患者预后更差[132];心电图碎裂QRS 波[133]、血浆内皮素水平升高[134]、血尿酸水平升高[135]、高敏C 反应蛋白升高[136]、女性[137]等指标与患者不良事件有关,但与SCD 的关系有待进一步明确。
6 治疗
HCM 治疗的总体原则是减轻症状,改善心功能,延缓疾病进展。由于HCM 发病机制主要是肌小节蛋白编码基因变异,因此常规药物很难从根本上解决心肌肥厚所导致的一系列临床症候群。对非梗阻性HCM 患者的治疗主要集中于控制心肌肥厚进展、降低左心室充盈压力、减轻临床症状,及治疗管理心律失常、心衰等合并症;对于梗阻性HCM 患者,可以通过药物、介入治疗、外科手术等来改善症状,降低风险。
6.1 非梗阻性HCM 治疗
无症状的非梗阻性HCM 患者,大多数是查体或无意中被发现。如仅表现为局部心肌肥厚,没有明显血流动力学改变,要启动临床观察和随访(Ⅱa,C),同时进行不良预后(如猝死)危险分层、合并症评估,如无禁忌,可适当选用药物治疗(如β 受体阻滞剂等)。
与无症状的HCM 患者相比,出现症状的HCM患者结局更差[138]。非梗阻性HCM 患者常见症状如呼吸困难和胸痛等,可能与心肌舒张功能障碍、肥厚的心肌需氧-供氧失衡、冠状动脉微血管受压或与其他合并症因素有关,如合并典型心绞痛症状或伴有冠心病多重危险因素的患者,应排除冠心病[68](Ⅰ,C)。合并心衰、心律失常等的非梗阻性HCM患者的治疗方案与无HCM 的心衰患者相似,应根据LVEF 进行分层,在健康生活方式的基础上,考虑合并疾病,进行个体化治疗(具体参阅心衰和心律失常治疗部分)(Ⅱa,C)。合并房颤的非梗阻性HCM 患者脑卒中风险增加,建议给予口服抗凝药物治疗,无需CHA2DS2-VASc 评分,启动抗凝治疗前建议进行出血评分[139](Ⅰ,B)。由于HCM 患者对快速室率的耐受性较差,维持窦性心律和控制心率是治疗的关键[21](Ⅰ,B)。
6.2 梗阻性HCM 治疗
与非梗阻性HCM 患者相比,左心室流出道梗阻的患者生存率较低,缓解梗阻可能有助于降低死亡风险[3]。
6.2.1 药物治疗
6.2.1.1 常规药物治疗
梗阻性HCM 药物治疗的主要目标是缓解症状。
(1)梗阻性HCM 患者,在无禁忌证情况下,根据心率、血压情况,从小剂量开始使用β 受体阻滞剂,逐步滴定至最大耐受剂量[140](Ⅰ,B)。除非在治疗过程中有明显β 受体阻滞剂不耐受。
(2)β 受体阻滞剂无效或不耐受的患者,可选用非二氢吡啶钙拮抗剂(如维拉帕米、地尔硫)(Ⅰ,B)。目前尚缺乏β 受体阻滞剂与非二氢吡啶钙拮抗剂联合使用治疗HCM 的证据,但这种联合可用于HCM 合并高血压的患者[2]。
(3)对于使用β 受体阻滞剂或非二氢吡啶钙拮抗剂后仍有明显症状的患者,可联用丙吡胺(Ⅰ,B)。由于丙吡胺可增强房室结传导,在房颤发作时有增加室率的可能,故建议应与β 受体阻滞剂或非二氢吡啶钙拮抗剂联合使用。
(4)梗阻性HCM 患者易因左心室负荷变化发生严重低血压,如果补液效果不佳,建议静脉注射苯肾上腺素或其他无正性肌力活性的血管收缩剂。β 受体阻滞剂可以延长舒张期充盈、抑制心肌收缩力[2,141],必要时可考虑联合使用(Ⅰ,C)。
(5)梗阻性HCM 合并持续性呼吸困难的患者,临床证据显示容量过载或左心室充盈压高时,可考虑使用小剂量口服利尿剂(Ⅱb,C),但过量利尿会降低前负荷而加重左心室流出道梗阻[2]。
(6)梗阻性HCM 患者,使用具有血管扩张作用的药物如血管紧张素转换酶抑制剂(angiotensinconverting enzyme inhibitor,ACEI)/血管紧张素Ⅱ受体拮抗剂(angiotensin Ⅱ receptor blocker,ARB)、二氢吡啶钙拮抗剂等,以及地高辛或大剂量利尿剂可能有害,原因与这些药物加重流出道梗阻有关[2](Ⅲ,C)。
(7)严重梗阻性HCM 患者,静息时LVOTG>100 mmHg,出现静息状态严重的呼吸困难、低血压时,维拉帕米可能有害(Ⅲ,C)。这类药物具有的血管扩张作用,使用后会加重低血压及LVOTG,增加风险[73]。
(8)左心室中部梗阻的HCM 患者药物治疗可以参考左心室流出道梗阻治疗建议,使用剂量宜个体化[142](Ⅱa,C)。
6.2.1.2 靶向药物治疗
2016 年Science 杂志发表了小分子化合物MYK-461 可以作为HCM 治疗的潜在药物研究,之后这个化合物被命名为Mavacamten。可以靶向作用于心肌肌球蛋白ATP 酶,减少肌动蛋白-肌球蛋白横桥的形成,从而减轻心肌的过度收缩,改善舒张功能。多中心临床研究显示Mavacamten 可降低LVOTG,改善心功能和症状[143-144]。因此,FDA 批准该药可用于NYHA 心功能分级Ⅱ~Ⅲ级且有症状的成人梗阻性HCM 患者。但因该药目前缺乏中国人的数据,本指南暂未形成具体的推荐类别和证据水平建议。
Aficamten(又称CK-274)是第二个进入临床试验的小分子肌球蛋白抑制剂,相比Mavacamten 其半衰期更短,可在2 周内达到稳定浓度,治疗窗相对更宽,Ⅲ期临床试验正在进行中[145]。
6.2.2 介入治疗
临床上主要包括经皮腔内室间隔心肌消融术(percutaneous transluminal septal myocardial ablation,PTSMA)、经皮心肌内室间隔射频消融术(percutaneous intramyocardial septal radiofrequency ablation,PIMSRA)和经皮心内膜室间隔射频消融术(percutaneous endocardial septal radiofrequency ablation,PESA)。
6.2.2.1 经皮腔内室间隔心肌消融术
PTSMA 是通过导管将无水酒精注入左前降支的一支或多支间隔支中,造成相应肥厚部分的心肌梗死,使室间隔基底部变薄,以减轻LVOTG 和梗阻的方法。PTSMA 对于有适应证的HCM 患者可有效降低LVOTG、改善症状,增加活动耐量,长期预后良好,恶性心律失常及猝死发生率无明显增加[146-147]。
(1)适应证:同时具备临床适应证至少一项、血流动力学适应证和形态学适应证的患者建议行PTSMA,并建议在三级医疗中心由经验丰富的团队进行[146-148](Ⅰ,C)。①临床适应证:a.经过规范药物治疗3 个月静息或轻度活动后仍出现临床症状,或有严重不良反应,基础心率控制在60 次/min 左右,NYHA 心功能分级 Ⅲ/Ⅳ级或加拿大心血管病学会(Canadian Cardiovascular Society,CCS)胸痛分级Ⅲ级;b.尽管症状不严重,NYHA 心功能分级未达到Ⅲ/Ⅳ级,但有其他猝死的高危因素,或有运动诱发的晕厥;c.外科室间隔切除术或植入带模式调节功能的双腔起搏器失败;d.有增加外科手术危险的合并症的患者。②血流动力学适应证:经胸超声心动图静息状态下LVOTG ≥50 mmHg,或激发后LVOTG ≥70 mmHg。③形态学适应证:a.室间隔厚度≥15 mm,梗阻位于室间隔基底段,且合并与SAM 征有关的左心室流出道及左心室中部压力阶差,排除乳头肌受累和二尖瓣叶过长;b.冠状动脉造影有合适的间隔支,间隔支解剖形态适合介入操作。心肌声学造影可明确拟消融的间隔支为梗阻心肌提供血供,即消融靶血管。
(2)禁忌证(Ⅲ,C):①非梗阻性HCM;②合并必须行心脏外科手术的疾病,如严重二尖瓣病变、冠状动脉多支病变等;③无或仅有轻微临床症状,无其他高危因素的患者;④不能确定靶间隔支或球囊在间隔支不能固定;⑤室间隔厚度≥30 mm,呈弥漫性增厚;⑥终末期心衰;⑦年龄虽无限制,但原则上对年幼患者禁忌,高龄患者应慎重;⑧已经存在左束支阻滞者。
6.2.2.2 经皮心肌内室间隔射频消融术
PIMSRA 是在超声实时引导下,在心脏非停跳状态下,将射频针经皮肤、肋间、心尖精准穿刺直接送至室间隔心肌肥厚部位,利用射频电极针前端发出的高频交变电流,使肥厚心肌组织局部升温、心肌细胞脱水,造成不可逆凝固性坏死;同时,可使消融心肌内间隔支发生凝固形成反应带,从而阻断肥厚心肌组织血供,最终使室间隔厚度变薄、狭窄处内径增宽,从而缓解梗阻。中短期随访显示,PIMSRA 可以有效减低室间隔厚度,降低左心室流出道或心腔内压力阶差,改善患者症状,提高患者的生活质量[149-151]。
(1)适应证:①经过最大耐受剂量药物治疗后,仍有显著呼吸困难、胸痛或有运动诱发的晕厥等严重临床症状,NYHA 心功能分级Ⅲ/Ⅳ级或CCS 胸痛分级Ⅲ/Ⅳ级;同时超声心动图检查静息或激发状态下LVOTG 或左心室腔内压力阶差≥50 mmHg。②尽管未满足上述条件,但有其他猝死的高危因素或严重症状的患者。
(2)禁忌证:①无症状非梗阻性HCM;②严重心衰患者,经强化抗心衰治疗,仍有静息性心衰症状,LVEF <35%;③近6 个月发生临床事件:心脏骤停抢救存活或植入ICD 适当放电、急性心肌梗死、因心衰住院、血栓栓塞等。
(3)特殊问题处理:合并冠状动脉狭窄需要血运重建治疗的患者,可于术后6 个月行PIMSRA 进行室间隔减容术。
PIMSRA 作为一种创新术式,建议在三级医疗中心由经验丰富的多学科团队(心脏超声介入医师、心外科、心内科)实施。其短-中期疗效良好,未明显增加远期心律失常风险[151-152],但相关经验和长期随访资料较为有限,本指南暂未形成具体的推荐类别和证据水平的建议。
6.2.2.3 经皮心内膜室间隔射频消融术
PESA 是利用心腔内三维超声导管同时将梗阻区和心脏关键传导束直接描绘到电生理三维标测图上,最大限度地避免消融时传导束的损伤。消融导管在梗阻室间隔区释放射频能量,使肥厚梗阻的室间隔短期内水肿,心肌顿抑,疤痕化后萎缩,随后心肌向心性收缩力激动顺序发生改变,这些综合因素使LVOTG 减低,缓解梗阻[153]。与外科切除和化学消融不同,PESA 是在不显著减轻肥厚心肌的基础上通过肥厚心肌疤痕化和收缩顺序改变来减低LVOTG。有荟萃分析比较了PESA 和外科切除术两者之间的疗效,发现在减小室间隔厚度方面,外科切除术明显优于PESA,而在改善心功能方面,二者类似[154]。该术式对梗阻性HCM 伴晕厥者疗效明确[155]。PESA的相关经验和长期随访资料较为有限,本指南暂未形成具体的推荐类别和证据水平的建议。
6.2.3 外科手术治疗
6.2.3.1 手术术式
(1)Morrow 手术:经典的室间隔肥厚心肌切除术。经主动脉切口,在主动脉瓣下5 mm 作两平行纵切口,第一切口从右冠瓣中点朝向心尖,第二切口从左右冠瓣交界朝向心尖;切除长度约2~3 cm,切除厚度约为室间隔厚度的50%[156]。缺点是左心室中部的室间隔肌肉切除不完全,流出道疏通不充分。
(2)改良扩大Morrow 手术:目前广泛采用。切除膜部室间隔以左3~5 mm 至接近二尖瓣前交界之间的肥厚心肌;切除范围向心尖延伸并超越左心室流出道梗阻最严重的部位后达到二尖瓣乳头肌根部水平,即长度扩大到5~7 cm。切除还包括左心室外侧壁、后壁连接处的心肌[157]。
(3)经二尖瓣口左心室腔中部梗阻疏通术:可通过常规方式、胸腔镜或机器人辅助腔镜微创技术实现[158-160]。对于合并有二尖瓣病变需要进行二尖瓣处理的患者尤其合适,但对不需要进行二尖瓣处理的患者会增加二尖瓣关闭不全的风险。这些新手术方式的有效性和安全性仍有待将来大规模临床验证。
(4)经心尖心肌切除术:适用于心尖肥厚为主或合并心尖肥厚导致左心室腔缩小和舒张功能不全的患者。在心尖处切开心室,切口一般长6 cm,选择在左前降支动脉外侧并需避免切口缝合时损伤该动脉,避免损伤乳头肌。沿室间隔切除肥厚的心肌组织,而左心室游离壁的心肌组织基本保留。如遇到特别肥厚的乳头肌且导致左心室腔中部梗阻,也可作适当切除。近年来,对于一些复杂的HCM 患者,有术者采用经主动脉联合心尖切口切除心肌,可以保证心室腔内任一水平的梗阻都可得到解除[161]。
(5)经右心室心肌切除术:对于合并右心室壁肥厚的HCM 患者,可在右心室圆锥部作2~3 cm 的切口,分离室间隔前部和右心室前壁的肌梁肉柱,并通过该切口切除室间隔肌肉,切除部位则和引起左心室流出道梗阻的部位相对应,厚度一般不超过室间隔厚度的1/2[157]。
6.2.3.2 适应证
室间隔心肌切除术推荐由经验丰富的外科医师实施[2]。
(1)对于NYHA 心功能分级Ⅲ/Ⅳ级,或有严重胸痛,或偶尔出现流出道梗阻导致的其他劳累症状(如晕厥、先兆晕厥)的流出道梗阻患者,尽管采用了合理的、最大耐受剂量的药物治疗,但仍影响到日常活动或生活质量;且静息或运动激发下的LVOTG ≥50 mmHg,与室间隔肥厚和二尖瓣SAM征有关;且拟切除的室间隔的厚度足以安全有效地进行手术(Ⅰ,B)。
(2)对于NYHA 心功能分级Ⅱ级的流出道梗阻患者,当合并以下任一种情况时(Ⅱb,B):①存在由流出道梗阻或因之发生的二尖瓣反流导致的严重进展性肺动脉高压;②左心房增大并发作过症状性房颤;③平板运动试验提示流出道梗阻导致的心功能低下。
(3)对于有症状的流出道梗阻患者,如同时合并其他相关需要手术干预的心脏疾病时,如异常的乳头肌、显著的二尖瓣前叶延长、二尖瓣本身病变、多支血管狭窄的冠心病、主动脉瓣狭窄等(Ⅰ,B)。
(4)对于静息或运动激发下LVOTG 30~50 mmHg 时,药物治疗反应不佳,症状明显的患者(Ⅱb,B)。
(5)如果患者室间隔肥厚严重(>30 mm),尽管LVOTG <50 mmHg 和症状不明显,若合并其它需要手术干预的心脏疾病,建议同时行室间隔肥厚心肌切除术(Ⅱb,B)。
6.2.3.3 禁忌证(Ⅲ,C)
(1)对于无症状的流出道梗阻HCM 患者,如日常活动耐量正常,不推荐行外科手术治疗。
(2)如果行室间隔心肌切除可以疏通流出道,而无二尖瓣器质性病变者,不推荐二尖瓣换瓣治疗。
(3)高龄、合并严重并发症,手术风险被认为不可接受的患者。
6.2.3.4 特殊问题处理
(1)二尖瓣异常:梗阻性HCM 多合并二尖瓣关闭不全,绝大多数不需要实施二尖瓣手术,解除梗阻后二尖瓣反流大部分可消除[162]。对于术前二尖瓣本身有病变的患者,在行肥厚心肌切除的同时干预二尖瓣及其附属器,包括成形和换瓣[163]、腱索切断[164]等(Ⅱa,B)。
(2)合并冠状动脉病变:对年龄≥50 岁、有胸痛症状或年龄≥40 岁伴有冠心病危险因素的患者常规行冠状动脉造影检查,若合并严重冠状动脉病变,建议在行肥厚心肌切除的同时行冠状动脉血运重建治疗(Ⅱa,B)。
(3)心肌桥:HCM 合并心肌桥发生率为15%~40%[165],多见于左前降支,如果考虑HCM 患者的胸痛等症状与心肌桥相关,可在肥厚心肌切除的同时切开肌桥位置冠状动脉表面的心肌或行冠状动脉旁路移植术[166](Ⅱb,B)。
(4)房颤:对于合并房颤的患者,建议在肥厚心肌切除的同时行房颤迷宫手术[167](Ⅱa,B)。
(5)乳头肌异常:有些HCM 患者还需要切除部分粗大的乳头肌,以解除心室腔中部的梗阻。如遇到异位乳头肌直接连接二尖瓣瓣体导致流出道梗阻,则需同期切除;但如果异位乳头肌连接的是二尖瓣瓣缘,则该乳头肌需要保留[156](Ⅱb,B)。
6.2.3.5 有效性和安全性
肥厚心肌切除外科手术的安全性已被证实,有经验的中心手术死亡率小于1%,成功率在90%~95%以上[168],术后远期生存率与年龄匹配的普通人群相近[169]。如同期行二尖瓣手术,围术期死亡率约为3%~4%[73],而二尖瓣成形相对瓣膜置换能提高患者生存率[163]。远期效果良好的决定因素是年龄<50 岁、左心房内径<46 mm、术前无房颤和男性患者[170]。
6.2.3.6 手术并发症
(1)传导阻滞:术后大约2%的患者出现完全性房室阻滞。完全性左束支阻滞发生率50%~76%,若术前患者存在完全性右束支阻滞,外科术后则更易发生完全性房室阻滞,需要植入永久性起搏器[171-172]。
(2)室间隔穿孔(<1%):多发生于室间隔厚度<18 mm 的患者。术中经食道超声心动图检查发现有室间隔左向右分流,在除外左心室冠状动脉瘘后,应及时进行手术修补[168]。
(3)主动脉瓣反流(<1%):多发生于低龄患者以及主动脉瓣环较小的患者,因主动脉瓣损伤所致。一旦发生应积极给予干预[158]。
(4)残余梗阻(2%):若术毕经食道超声心动图检查发现LVOTG>30 mmHg,应再次进行手术处理[169]。
6.2.4 植入双腔起搏器
起搏器的原理是使用短的AV 间期改变了左心室的激动顺序,远离肥厚室间隔部位的心肌提前激动和收缩,而肥厚室间隔的激动和收缩相对滞后,随之减轻左心室流出道梗阻。对梗阻性HCM 患者植入起搏器需注意两点:(1)心室起搏电极必须置于真正的右心室尖部。(2)房室间期必须短于患者窦性心律的PR 间期。起搏治疗的疗效与选择合适的房室间期有关。
对于部分静息或激发时LVOTG ≥50 mmHg、窦性心律且药物治疗无效的患者,若合并介入治疗或外科手术治疗禁忌证,或术后发生心脏传导阻滞风险较高,应考虑植入双腔起搏器,通过房室顺序起搏并优化房室间期,以降低LVOTG,并改善药物疗效[173](Ⅱb,B)。
6.3 合并心衰的治疗
6.3.1 射血分数保留的心衰
HCM 表现为HFpEF 的患者,既有心肌被动充盈障碍也有主动舒张障碍,因此治疗重点涵盖这两部分内容。
(1)改善被动充盈障碍:HCM 患者被动充盈障碍的主要发病机制类似于高血压、冠心病等所致舒张功能不全,目前临床常规药物主要是针对这一机制。①β 受体阻滞剂:减慢心率,延长心室舒张期,降低心肌收缩力,降低心肌耗氧,从而改善心室功能。β 受体阻滞剂是HCM 改善症状一线治疗药物(Ⅰ,B),可选用美托洛尔、比索洛尔。应从小剂量起始,逐渐增加至最大耐受剂量(患者能够耐受情况下静息心率达到55~60 次/min)。普萘洛尔最早应用于HCM,目前仍可应用,起始10 mg/次,3~4次/d,逐渐加量,最大可达每日200 mg。另外阿替洛尔和索他洛尔也可用于HCM[174]。②非二氢吡啶类钙拮抗剂:具有负性肌力和负性频率作用,可以改善心室舒张期充盈和局部心肌血流。对于β 受体阻滞剂治疗有禁忌或不能耐受的患者,可以应用维拉帕米(Ⅰ,B)(起始40 mg/次,3 次/d,最大剂量每日480 mg)。对于β 受体阻滞剂及维拉帕米不能耐受或存在禁忌患者,可应用地尔硫(Ⅱa,C)(起始60 mg/次,3 次/d,最大剂量每日360 mg)。③利尿剂:由于HCM 患者心肌肥厚导致的左心室舒张末期容积减小,每搏量减少,使用利尿剂会改变心室容积导致搏出量的很大变化,因此使用时应注意避免负荷过度降低而导致的低血压[175]。对有心衰症状的HCM 患者,在心率控制基础上血压能耐受的情况下可使用小剂量利尿剂(Ⅱa,C)。
(2)改善主动舒张障碍:主动舒张障碍是HCM的主要病理生理改变,常规药物对此作用有限。心肌肌球蛋白ATP 酶抑制剂Mavacamten 的上市为该类患者的主动舒张障碍治疗提供了一种选择,具体内容如前述。
6.3.2 射血分数降低的心衰
HCM 通常被排除在心衰相关的随机对照试验之外,迄今缺乏充分的证据表明HCM 患者出现射血分数降低的心衰(heart failure with reduced ejection fraction,HFrEF)与其它病因的HFrEF 在治疗上存在差异,因此治疗基本等同[175-176](Ⅰ,C)。
(1)β 受体阻滞剂:推荐长期应用β 受体阻滞剂(美托洛尔、比索洛尔、卡维地洛等),能改善症状和生活质量,降低死亡、住院、猝死风险,除非有禁忌证或不能耐受[177](Ⅰ,A)。
(2)肾素-血管紧张素系统抑制剂:无严重流出道梗阻者推荐应用ACEI[178](Ⅰ,A)、ARB[179](Ⅰ,A)或血管紧张素受体脑啡肽酶抑制剂(angiotensin receptor-neprilysin inhibitor,ARNI)(Ⅰ,B)[180]抑制肾素-血管紧张素系统,可降低心衰的死亡率。
(3)醛固酮受体拮抗剂:推荐在使用ACEI/ARB/ARNI、β 受体阻滞剂的基础上加用醛固酮受体拮抗剂(Ⅰ,A),可使NYHA 心功能分级Ⅱ~Ⅳ级的HFrEF 患者获益,降低全因死亡、心血管死亡、猝死和心衰住院风险[181]。
(4)利尿剂:利尿剂的应用可以减轻水钠潴留,有效缓解心衰患者的呼吸困难及水肿,改善运动耐量(Ⅰ,C)。
(5)钠-葡萄糖共转运蛋白2 抑制剂:无论是否合并糖尿病,HFrEF 患者应用达格列净或恩格列净可以改善患者临床症状与生活质量,降低因心衰住院和(或)减少心血管死亡[182]。
6.4 合并心律失常的治疗
6.4.1 合并房性心律失常
心肌肥厚导致心室舒张功能受损,心房张力增加,肾素-血管紧张素-醛固酮系统激活,心房纤维化等因素,均是HCM 患者易发生房性心律失常尤其是房颤的原因,房颤的发生率约20%~25%[183-184],在梗阻性和老年HCM 中发生率更高。合并房颤比不合并房颤的HCM 患者死亡率增高4 倍[20]、脑卒中风险增加8 倍[185-186]。HCM 合并其他室上性心律失常和房扑的发生率与非HCM 的人群可能相似[187]。
6.4.1.1 合并房颤的药物治疗
抗凝治疗:荟萃分析显示,HCM 伴房颤患者的血栓栓塞患病率为27%[186]。心脏植入设备或监护仪检测到的无症状房颤也会增加脑卒中风险,因此抗凝治疗应考虑发作的持续时间以及潜在的危险因素以及合并症等。
对于HCM 合并临床房颤的患者,无论CHA2DS2-VASc 评分情况,在无禁忌证时均建议抗凝治疗。除非房颤病因可逆转,否则在恢复窦性节律前建议口服抗凝治疗。合并房扑时按房颤进行抗凝治疗。建议使用直接口服抗凝剂(direct oral anticoagulant,DOAC)作为一线选择,维生素K 拮抗剂作为二线选择[139,188](Ⅰ,A)。对于亚临床房颤的HCM 患者,若发作持续时间超过24 h,无论CHA2DS2-VASc 评分情况,也建议抗凝治疗。建议使用DOAC 作为一线选择,维生素K 拮抗剂作为二线选择[188](Ⅰ,B)。对于亚临床房颤的HCM 患者,若发作持续时间>5 min 但<24 h,考虑到房颤发作的持续时间、总房颤负担、潜在危险因素和出血风险,以DOAC 作为一线选择和维生素K 拮抗剂作为二线选择的抗凝治疗可能是有益的(Ⅱa,C)。
HCM 人群中脑卒中的危险因素包括高龄、既往栓塞事件、NYHA 心功能分级、左心房内径、血管疾病和最大左心室壁厚度[189],合并这些危险因素的HCM 人群注意房颤的识别。当观察到非常短的房颤发作时,应继续监测,因为房颤的负担可能会加重。
抗凝治疗前,应考虑采用HAS-BLED 评分评估出血风险[188](Ⅱa,B)。
6.4.1.2 节律和室率控制
HCM 患者对房颤的耐受性较差,节律控制优于室率控制[21](Ⅰ,B)。
(1)药物治疗:①对采用节律控制策略的患者,如果LVEF<50%,胺碘酮是首选,可以合并使用β受体阻滞剂、ACEI、ARB、螺内酯、袢利尿剂[73](Ⅱa,C)。口服或静脉注射用索他洛尔也是安全的,通常用于儿童[190](Ⅱa,C)。丙吡胺虽然已安全地用于左心室流出道梗阻患者,但其在房颤中的疗效尚不确定[191]。ⅠC 类抗心律失常药物的使用尚有争议,一般情况下心律平禁用,该药可能使房颤转为房扑,出现1∶1 下传,引发急性恶性血流动力学改变(Ⅲ,B)。②对于采用心室率控制策略的患者,推荐使用β 受体阻滞剂、维拉帕米或地尔硫,并根据患者偏好和共病情况选择药物(Ⅰ,C)。剂量调整基于充分的心室率控制与副作用(包括低血压及严重的心动过缓)之间的平衡。在无左心室流出道梗阻的情况下,地高辛是一种潜在的选择(Ⅱa,C)。③急诊处置:对于HCM 发作房颤时出现急性血流动力学不稳定(如低血压、急性左心衰、晕厥/先兆晕厥)者,体外直流电复律是首选(Ⅱa,C)。复律后再口服胺碘酮维持窦性心律是合理的[73](Ⅱa,B)。对于血流动力学稳定患者,首次发作房颤时可静脉使用胺碘酮复律(Ⅰ,A)。
(2)介入治疗:经导管消融(射频或冷冻)治疗是房颤节律控制的一线治疗,优于单纯的药物治疗。这一方面是因为部分HCM 患者在房颤发作前常合并窦性心动过缓,房颤发作时无论是用药物进行节律控制还是室率控制都有顾虑;另一方面HCM 患者在合并房颤时往往血流动力学和心功能会急剧恶化,也使药物复律受限,而导管消融可快速解除症状,又能减少药物使用负荷。因此,导管消融成为HCM 合并症状性房颤和不耐受药物者的一个选择(Ⅱa,B)。荟萃分析显示HCM 房颤患者,一次射频消融后1 年窦性心律维持率45.5%,二次或多次射频消融术后6 年窦性心律维持率61%,但需要配合抗心律失常药维持[192]。
对于左心房巨大(左心房内径>50 mm)、长时程房颤、LVEF<50%、NYHA 心功能分级Ⅲ/Ⅳ级,节律控制和室率控制都难以实现者,直接消融房室结后植入心脏起搏器是可行的[73](Ⅱa,C)。
6.4.2 合并室性心律失常
HCM 患者常合并室性期前收缩、NSVT,也易发生多形性室速及室颤。HCM 患者发生单形性室速少见,多与合并室壁瘤相关。HCM 患者易发生SCD,其原因与室性心律失常密切相关[193]。药物治疗的主要目的是减少室性心律失常,改善症状,并提高患者的生活质量。抗心律失常药物预防SCD 的疗效有限。
6.4.2.1 药物治疗
(1)如无禁忌证,无血管扩张作用的β 受体阻滞剂应作为首选治疗药物,并逐渐加量至最大耐受剂量(Ⅰ,B)。
(2)对于尽管服用了足量的β 受体阻滞剂或非二氢吡啶类钙拮抗剂,但仍出现有症状的室性心律失常,或植入ICD 后反复发生电击治疗的患者,建议使用胺碘酮(Ⅰ,B)、美西律(Ⅰ,C)或索他洛尔治疗(Ⅰ,C)。选择药物时结合年龄、疾病严重程度、合并症、患者偏好以及疗效和安全性之间的平衡综合考虑[194-195]。
(3)对于有持续性室速或室颤病史的患者,如果ICD 植入不可行或患者不愿接受ICD,使用胺碘酮可能有益[194](Ⅱb,B)。
由于HCM 患者室速管理的数据有限,大多数关于室速二级预防的研究都是从对非HCM 患者的研究中推断出来的。胺碘酮可能最有效,但副作用增加[194]。美西律的疗效证据很少,但通常是作为胺碘酮的辅助药物[195]。ⅠC 类药物普罗帕酮和氟卡胺的安全性和有效性尚不确定,并且在缺血性心脏病患者中使用时存在安全性顾虑[196]。
6.4.2.2 器械治疗
器械治疗手段包含两类:ICD 和导管消融治疗。目前认为预防HCM 患者SCD 的可靠方法只有植入ICD[73]。导管消融治疗仅作为减少室性心律失常发作的辅助手段。鉴于在60 岁以上HCM 患者中观察到的SCD 事件率较低,现有的SCD 一级预防策略最适用于青年和中年HCM 患者[197]。此外,对于符合SCD一级预防策略的患者,应在患者充分了解风险获益比的情况下,与患者共同商讨制定ICD 相关的决策。

HCM 器械治疗推荐建议
6.5 终末期治疗
6.5.1 心脏移植
心脏移植是HCM 终末期治疗最有效的手段。美国器官捐献共享网络登记数据显示,HCM 患者接受心脏移植术后1 年生存率为91.6%、5 年生存率为82.5%,总体生存情况优于缺血性心肌病(1 年生存率87.5%、5 年生存率75.3%)和其他类型非缺血性心肌病患者(1 年生存率91.3%、5 年生存率77.2%)[206]。中国医学科学院阜外医院数据显示HCM 心脏移植术后1 年生存率为89.5%、5 年生存率为83.7%,生存情况与国际水平相当[207]。影响HCM 患者心脏移植术后生存的主要术前因素包括:肺动脉高压、eGFR<60 ml/(min·1.73m2)、肾功能衰竭需要透析治疗、总胆红素升高等[206]。心肺运动试验是心脏移植受者筛选的重要评估手段,对HCM危险分层和预后判断具有很好的价值[208]。因此,HCM 患者在随访过程中,应对肺动脉高压、肾功能不全等危险因素进行识别,必要时对非梗阻性HCM及时进行心肺运动试验,以避免丧失心脏移植机会或严重影响心脏移植术后生存。
需要注意的是,临床上表现为心肌肥厚的终末期心脏病患者均需按照前文相关章节进行鉴别诊断,如淀粉样变、法布雷病、Danon 病等。
6.5.2 左心室辅助装置
由于HCM 患者左心室扩大不明显,LVEF 相对保留,传统观念认为左心室辅助装置并不适合HCM患者。然而,近年来一些病例系列报道显示,左心室舒张末期内径>50 mm 的HCM 患者可能从左心室辅助装置植入中获益[2],与扩张型心肌病患者相比,HCM 患者左心室辅助装置术后总体生存率、脑血管不良事件发生率、右心衰竭发生率、心律失常发生率和主动脉瓣反流发生率较高,但差异无统计学意义[209]。有限的文献显示,经过谨慎选择的部分HCM 患者可选择左心室辅助装置做为心脏移植术前的过渡治疗。
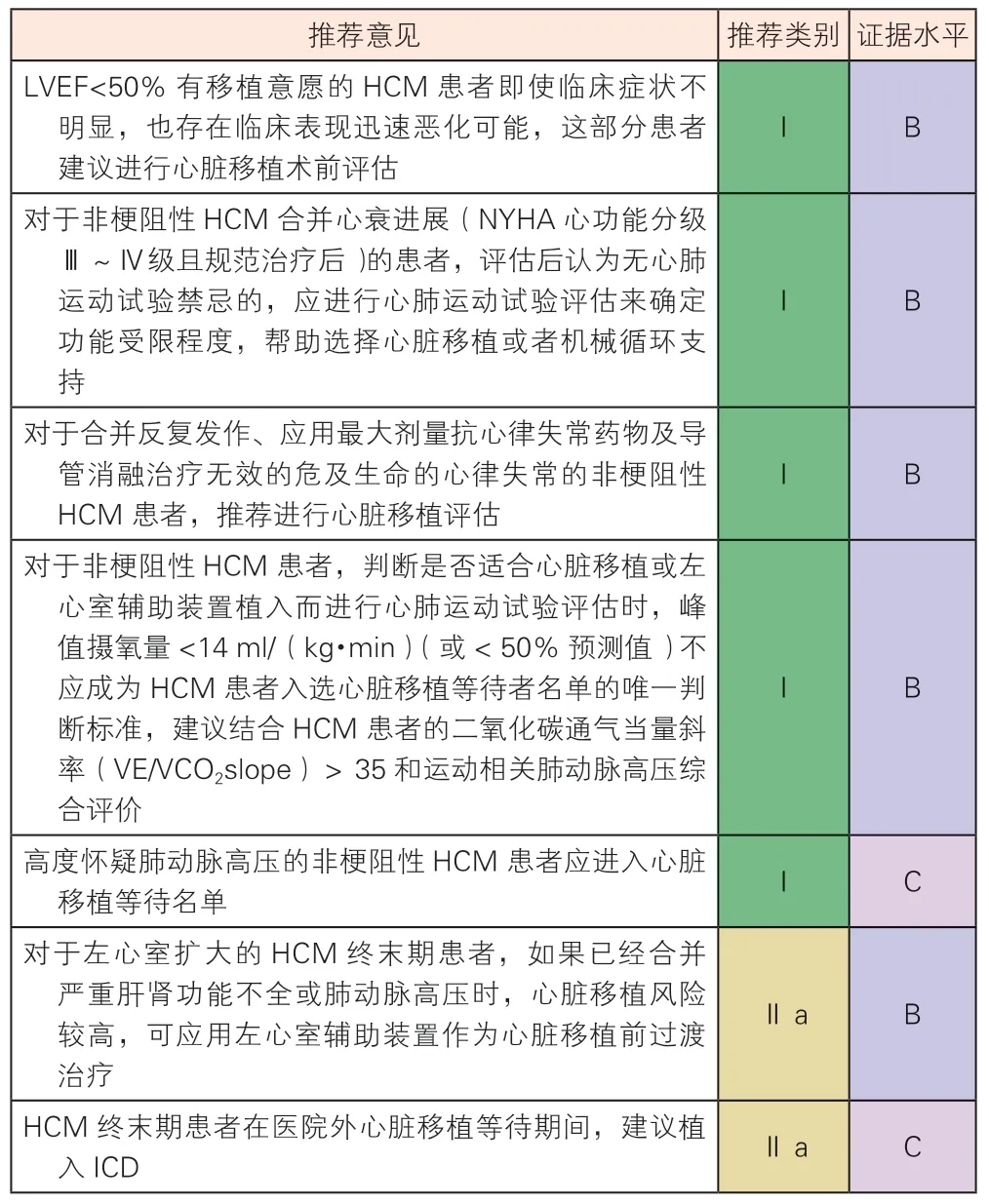
HCM 心脏移植及左心室辅助装置推荐建议
7 生活管理和随访
7.1 生活方式改变
改变生活方式即使不能治愈HCM,但依旧可以改善患者的健康程度和预期寿命。
7.1.1 运动
中等强度的个体化运动已被证明可以改善HCM患者健康相关客观指标,包括心率恢复、以代谢当量衡量的功能能力、运动时间、以峰值摄氧量衡量的运动能力及主观的临床症状和身体机能[210-211]。对于病情恶化和活动能力下降的HCM 患者,可通过增加肌力和耐力来提高日常生活能力和生活质量(Ⅱb,C)。
如无SCD 风险因素,可以参加低强度的运动和娱乐活动[212](Ⅱa,C)。如果需要,由多学科团队对参加体育活动的潜在风险进行全面评估和共同讨论后,可考虑参加高强度运动/竞技性运动(晕厥的发生可能与外伤或死亡有关的情况除外)[213-214](Ⅱb,C)。如果有任何风险增加的迹象,可以考虑参加低或中等强度的休闲运动[211,215](Ⅱb,C)。如果需要,HCM 基因型阳性但表型阴性的个体可以考虑参与所有运动,但应每年进行表型特征和风险分层评估(Ⅱb,C)。
7.1.2 饮食
HCM 患者应当均衡饮食,将体重指数保持在合适范围(Ⅰ,C),摄入不饱和脂肪酸而非饱和脂肪酸,减少盐的摄入量,多选择植物类的食物等[216-217]。
进食后会引起梗阻性HCM 患者发生心绞痛、呼吸困难、偶发晕厥等症状。建议HCM 患者少食多餐,减少餐后的即刻活动,保持出入量基本平衡[218](Ⅱa,C)。
不建议HCM 患者饮酒,尤其是梗阻性HCM 患者,防止流出道梗阻加重或隐匿梗阻的患者出现梗阻[219](Ⅲ,C)。
7.1.3 睡眠呼吸障碍
阻塞性睡眠呼吸暂停在HCM 中患病率为32%~71%[220]。对高风险的患者进行睡眠呼吸监测(Ⅱb,C),并积极治疗,可改善HCM 患者的预后。
7.1.4 心理支持
应进行定期心理筛查和咨询,特别是对于得知疾病遗传而懊悔或者对基因型阳性但表型阴性感到焦虑的患者[221],应正视其疾病并配合心理治疗(Ⅰ,C)。
7.1.5 就业
大多数HCM 患者能够继续正常的工作生活。经过临床综合评估,HCM 患者可以考虑从事需要体力劳动、提重物或需要较高强度体力活动的工作,但从事重体力劳动(如建筑工作)或高水平体力活动的职业(如执法人员、消防人员)可能给HCM 患者及公众带来风险,医护工作者与患者沟通时建议告知患者与职业要求相关的健康风险是不确定的[222](Ⅱb,C)。对于未植入ICD 且无SCD 主要风险因素的HCM 患者,可以考虑从事车辆驾驶(Ⅱa,C)。
7.1.6 旅行
对于无症状或轻度症状的HCM 患者,空中旅行是相对安全的。然而,航行时间长、高海拔、炎热和潮湿地区旅行仍需谨慎[221](Ⅱb,C)。
7.2 随访
随访是HCM 管理的重要部分。

HCM 随访推荐建议
8 妊娠
8.1 妊娠时HCM 的诊治
大部分HCM 的女性能够耐受妊娠,HCM 孕产妇死亡率非常低,妊娠前存在症状和心血管相关疾病的高危患者死亡风险增加。心衰和心律失常通常发生在妊娠晚期或产后[223],而合并左心室流出道梗阻的妊娠女性并未增加不良结局[2]。

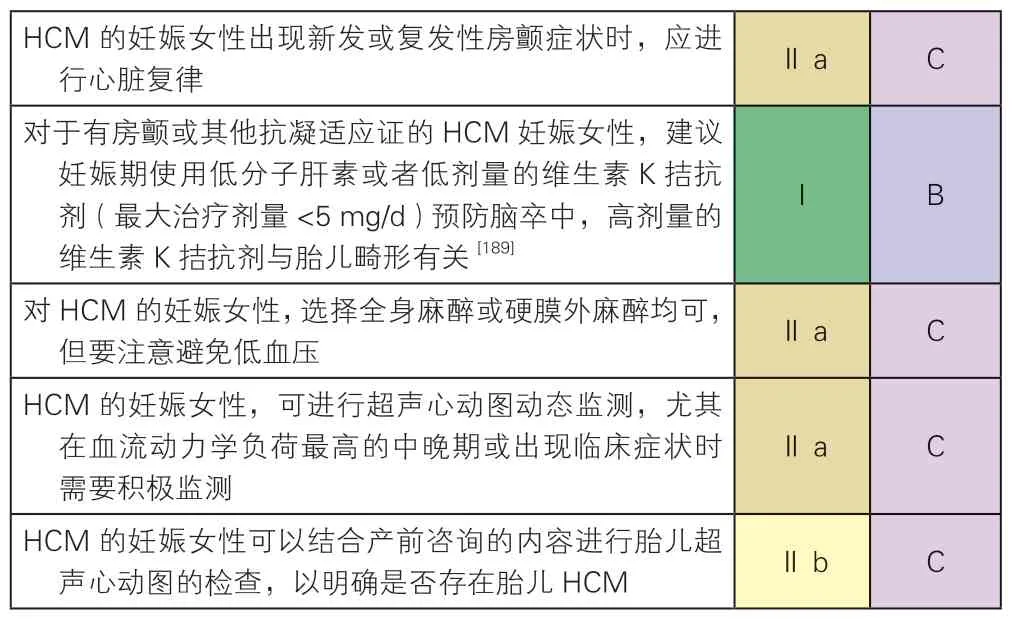
HCM 妊娠相关诊治问题的推荐建议
8.2 遗传阻断和选择性生育
HCM 患者的孕前遗传学检测十分必要。目前实验室基本采用候选基因筛查策略,具体参见基因诊断部分。基因检测结果的准确解释至关重要,未检测出致病变异并不能排除遗传疾病的可能性。
产前遗传咨询有助于解释疾病遗传的风险,并指导后续的生育选择。通过讨论每种选择的益处和潜在风险,个人或夫妇充分了解产前基因检测和胎儿筛查结果后做出最终决定[2]。准父母需要了解他们的后代遗传HCM 的可能,可以考虑自然受孕或其他方法来避免致病基因变异的遗传,其中包括产前诊断技术或胚胎着床前遗传学检测、配子捐赠(卵母细胞或精子)等。常规用于产前诊断的技术是绒毛膜绒毛取样或羊膜穿刺术。如果胎儿诊断为患病,父母可以在充分知情的情况下选择终止妊娠[224]。
胚胎植入前遗传学检测(preimplantation genetic testing,PGT)是通过一级预防策略实现HCM 的孕前阻断,降低HCM 患儿出生率的有效方式。PGT技术可检测胚胎是否携带相应HCM 基因变异以及染色体异常。囊胚滋养外胚层细胞活检的安全性和准确性较高,是目前临床主要活检方式。PGT 可分为以下三类:PGT-A、PGT-M 和PGT-SR,其中PGT-M 是针对单基因病胚胎着床前遗传学检测,而PGT-A 和PGT-SR 是分别针对着床前胚胎染色体数目和结构异常的检测。PGT-M 助孕的流程主要包括:(1)遗传和生育咨询;(2)夫妻双方进入常规辅助生殖治疗周期;(3)体外受精及胚胎活检;(4)胚胎遗传学检测;(5)胚胎移植;(6)产前诊断(例如羊水穿刺),降低胚胎诊断过程中误诊的风险;(7)新生儿随访[225]。高通量测序技术的应用可同时完成胚胎基因变异、染色体非整倍体以及家系连锁分析[226]。
对于同时满足以下情况的患者或家庭可建议其通过PGT 技术进行助孕,实现孕前HCM 的阻断:(1)临床诊断为HCM 的夫妇或生育过HCM 患儿的家庭;(2)已完成遗传学基因检测,基因致病位点诊断明确;(3)充分接受遗传咨询,知晓HCM 遗传特征、诊疗进展及生育风险,PGT 助孕的流程、局限性及风险等,患者知情同意选择[225,227]。当女方为HCM患者时,在孕前应对其出现的症状或表现出的高风险特征进行心内科、超声科、产科等多学科会诊,综合评估其身体状况能否妊娠、妊娠的风险、孕期管理及应对方案[225]。
9 多学科合作
通过多学科合作的综合方式对HCM 进行全面诊疗评估,有助于为患者选择个体化的诊疗方案,并获得最佳效果。
(1)经初级首诊机构的初步评估、治疗和护理后的具有心肌肥厚表现的患者,对于存在可优化预后的替代方案、对治疗决策有疑问或者需要进行侵入性治疗时,建议可考虑转诊至具有专业多学科HCM 团队的上级医疗机构[228-229](Ⅰ,C)。对各级医疗中心HCM 诊治中心建设要求可参考表2。
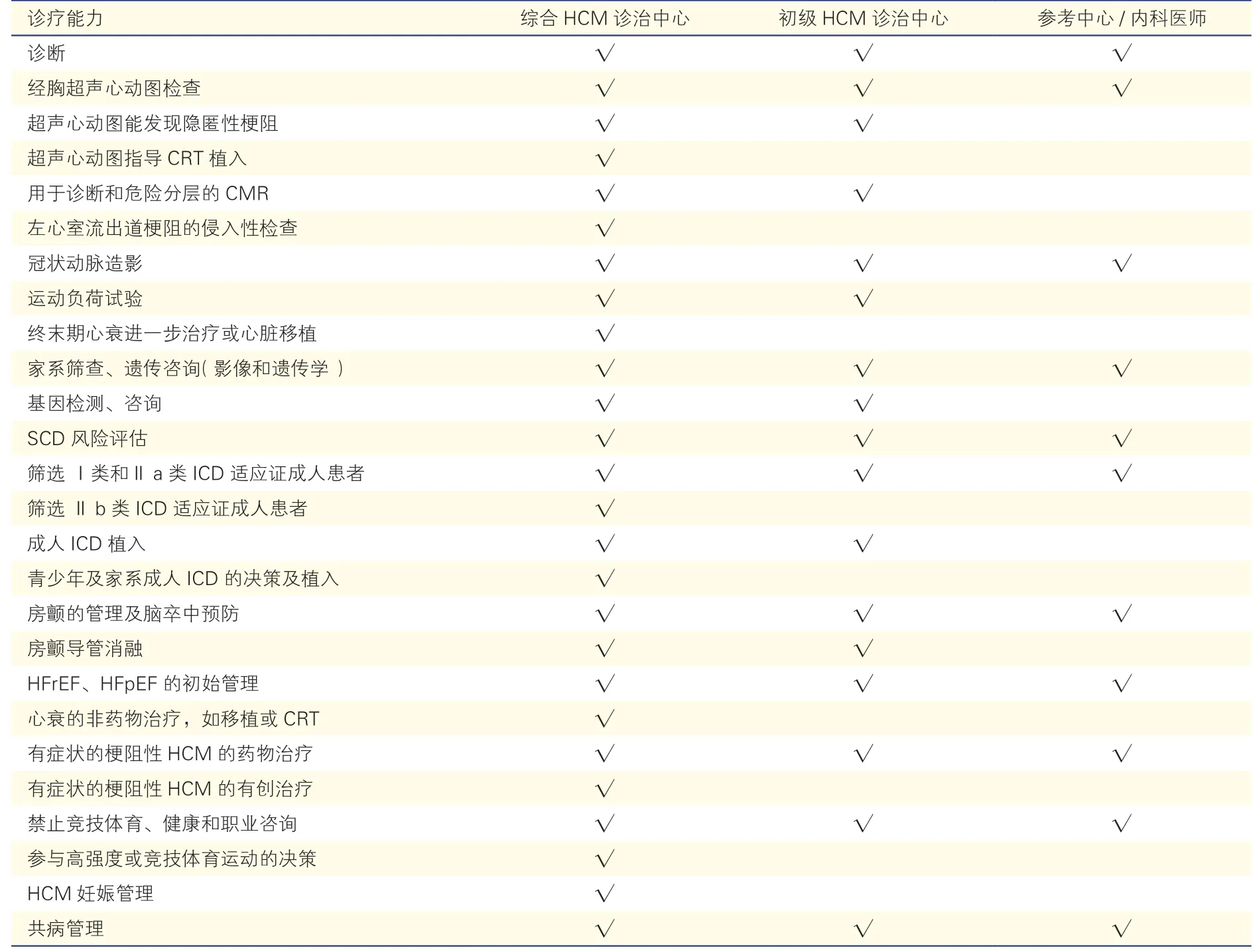
表2 各级HCM 诊治中心能力评价表
(2)需要进行鉴别诊断的具有心肌肥厚表现的患者,建议根据病史、临床表现、体格检查及辅助检查结果,联合心内科、血液科、肾内科、神经内科及内分泌科对各科疾病谱的系统性表现进行评估和鉴别[56],联合影像科(超声心动图、心肌核素显像及CMR)团队解读相关的影像学检查,联合病理科判断心肌活检结果等[230],进行综合性的精准诊断;对特定病因及系统性疾病所导致的HCM,在后续的治疗中,与患者及相关科室进行共同决策的治疗指导。
(3)对于出现多种并发症的HCM 患者,建议多学科团队(心内科、心外科、麻醉科等)的参与(Ⅱa,C):包括预防和治疗可能加重HCM 病情的合并症(如冠状动脉病变、肥胖、高血压、糖尿病、睡眠呼吸障碍等)[231-232];制订左心室流出道梗阻、心律失常及心衰等并发症的治疗方案(如室间隔减容术、导管消融术、ICD、心脏移植等),尤其在综合性HCM 中心可有助于做出复杂病况的外科手术相关管理决策。
(4)共同参与患者对治疗进行风险评估及预后的咨询。
(5)对患有HCM 的妊娠女性,由于合并妊娠的复杂性及各种生理学影响因素,包括妇产科的多学科会诊团队应在妊娠期全过程参与病情判断和诊疗;指导对有风险的妊娠女性进行产前检查和PGT。
(6)HCM 患者的运动处方应结合心内科及康复医学/运动医学科的参与,结合个体SCD 等危险因素和治疗目标,就训练强度以及参与的潜在风险进行全面讨论[233]。
(7)遗传/家系咨询是HCM 管理重要的一环,基因检测结果的解读建议由专业的心脏遗传咨询医师或具有心血管疾病遗传学知识的多学科团队进行系统地检测前和检测后遗传咨询[56](Ⅱa,C),包括提供关于疾病的遗传特点、识别高危人群以及对具有风险的亲属定期进行心血管评价。
指南指导专家组(按姓名汉语拼音排序):陈义汉(同济大学附属上海东方医院),杜杰(首都医科大学附属北京安贞医院),葛均波(复旦大学附属中山医院),韩雅玲(中国人民解放军北部战区总医院),胡盛寿(中国医学科学院阜外医院),惠汝太(中国医学科学院阜外医院),霍勇(北京大学第一医院),廖玉华(华中科技大学同济医学院附属协和医院),刘文玲(北京大学人民医院),马爱群(西安交通大学医学院第一附属医院),马长生(首都医科大学附属北京安贞医院),乔树宾(中国医学科学院阜外医院),汪道文(华中科技大学同济医学院附属同济医院),杨杰孚(北京医院),张抒扬(北京协和医院),张运(山东大学齐鲁医院)
指南写作组(按姓名汉语拼音排序):陈瑞珍(复旦大学附属中山医院),陈晓平(四川大学华西医院),陈旭华(中国医学科学院阜外医院),董建增(首都医科大学附属北京安贞医院),杜昕(首都医科大学附属北京安贞医院),方纬(中国医学科学院阜外医院),冯雪(中国医学科学院阜外医院),华伟(中国医学科学院阜外医院),黄洁(中国医学科学院阜外医院),贾玉和(中国医学科学院阜外医院),金玮(上海交通大学医学院附属瑞金医院),康连鸣(中国医学科学院阜外医院),李建平(北京大学第一医院),李新立(南京医科大学第一附属医院),李宗哲(华中科技大学同济医学院附属同济医院),刘金秋(大连医科大学附属第一医院),刘丽文(空军军医大学第一附属医院),罗晓亮(中国医学科学院阜外医院),区景松(中山大学附属第一医院),宋雷(中国医学科学院阜外医院),田庄(中国医学科学院北京协和医院),汪芳(北京医院),王红月(中国医学科学院阜外医院),王继征(中国医学科学院阜外医院),王曙霞(中国人民解放军总医院),王水云(中国医学科学院阜外医院),魏瑗(北京大学第三医院),肖红艳(武汉亚洲心脏病医院),徐希奇(北京协和医院),闫丽盈(北京大学第三医院),杨旗(首都医科大学附属北京朝阳医院),杨伟宪(中国医学科学院阜外医院),于汇民(广东省人民医院),袁建松(中国医学科学院阜外医院),曾春雨(陆军军医大学大坪医院),赵世华(中国医学科学院阜外医院),邹玉宝(中国医学科学院阜外医院)
指南学术秘书组(按姓名汉语拼音排序):蔡爽(中国人民解放军总医院),陈燕佳(上海交通大学医学院附属瑞金医院),陈志高(阜外华中心血管病医院),郭晓娟(首都医科大学附属北京朝阳医院),简宇鹏(中山大学附属第一医院),刘婕(中国医学科学院阜外医院),刘璐(四川大学华西医院),宋雅(中国医学科学院阜外医院),拓胜军(空军军医大学第一附属医院),汪蕾(中国医学科学院阜外医院),王宇成(复旦大学附属中山医院),王智(北京大学第一医院),魏之瑶(中国医学科学院阜外医院),吴桂鑫(中国医学科学院阜外医院),谢志鑫(广东省人民医院),张禅那(中国医学科学院阜外医院),张晔(陆军军医大学大坪医院),张志鹏(大连市中山医院),郑旭辉(南京医科大学第一附属医院),钟优(北京医院),朱昌盛(中国医学科学院阜外医院)
利益冲突:所有编写人员均声明不存在利益冲突
猜你喜欢
昆明医科大学学报(2021年4期)2021-07-23
宁夏医学杂志(2020年3期)2021-01-21
中国生殖健康(2020年7期)2020-12-10
江苏卫生保健(2020年9期)2020-10-23
天津医科大学学报(2019年6期)2019-08-13
中国临床医学影像杂志(2019年1期)2019-04-25
中西医结合心脑血管病杂志(2016年20期)2016-03-01
医学研究杂志(2015年4期)2015-06-10
中国当代医药(2015年30期)2015-03-01
郑州大学学报(医学版)(2015年2期)2015-02-27
