
高效液相色谱法同时测定蜈蚣药材中5种成分含量
2023-02-11 09:49马云马庆文范建伟李倩申凤霞关永霞张贵民
科学技术与工程 2023年1期
马云, 马庆文, 范建伟, 李倩, 申凤霞, 关永霞, 张贵民
(1.鲁南厚普制药有限公司, 临沂 276006; 2.中药制药共性技术国家重点实验室, 临沂 276006; 3.鲁南制药集团股份有限公司, 临沂 276006)
蜈蚣最开始记载于中国传统医药经典《神农本草经》,在中国有两千多年的药用历史,是临床上不可或缺的传统中药材[1]。《中国药典》中规定蜈蚣药材为蜈蚣科动物少棘巨蜈蚣ScolopendrasubspinipesmutilansL.Koch的干燥虫体,其主要功效为息风镇痉、通络止痛、攻毒散结,现代药理研究表明蜈蚣有镇痛、抗炎、抗菌、抗肿瘤、抗凝血、抗惊厥、抗凝纤溶、调节免疫、降压降脂等药理作用,在现代医学可用于治疗多种外科炎症、恶性肿瘤、糖尿病、癫痫、类风湿性关节炎、多种神经疼痛、血管栓塞性疾病,还可治疗百日咳、神经头痛、结核病等疾病[2-4]。众所周知,动物类药材有效物质基础不太明确,蜈蚣药材的研究同样难以避免此难题[5]。现行《中国药典》中对蜈蚣药材的质量控制方法较为简单,仅从性状、浸出物等方面对其进行鉴别评估,难以对药用蜈蚣药材质量进行有效评价,对蜈蚣药材开展多手段、多指标的质量控制方法研究,有利于完善其质量标准体系,保障药材质量和临床用药的安全有效[6]。化学成分是中药材起效的物质基础,从化学分子层面对蜈蚣药材展开多指标的质量评价研究,有利于完善其质量标准体系,更好地实现并保障中药材品质评价为临床疗效服务的最终目的[7]。经文献调研,尚未有蜈蚣药材多成分含量同时测定且基于含量测定对蜈蚣药材进行质量控制的相关研究,核苷类成分是生物细胞维持生命活动的基本组成物质,在动物、植物和微生物等生物中广泛存在,具有多种生理功能和药理活性[8]。如张琪等[9]发现蜈蚣中含有次黄嘌呤和黄嘌呤,次黄嘌呤和黄嘌呤具有抗肿瘤作用;3,8-二羟基喹啉对多种肿瘤细胞具有生长抑制及促凋亡的作用,能显著抑制脑神经胶质瘤细胞的体外增殖和侵袭迁移的作用[10-11];课题组从蜈蚣中分离得到硫酸-3-羟基喹啉、硫酸-3-羟基-4-甲氧基喹啉、3,8-二羟基喹啉[12];因此将次黄嘌呤、黄嘌呤、硫酸-3-羟基喹啉、硫酸-3-羟基-4-甲氧基喹啉、3,8-二羟基喹啉作为质量评价指标,旨在为蜈蚣药效物质基础的活性研究以及动物药中有效成分定性、定量分析提供参考。现通过高效液相色谱法建立蜈蚣中5种成分的含量测定方法,以期为蜈蚣药效物质基础的活性研究以及动物药中有效成分定性、定量分析提供参考。
1 材料
1.1 试剂
乙腈、甲醇(色谱纯,赛默飞世尔科技有限公司),异丙醇、磷酸(色谱纯,南京化学试剂股份有限公司),甲醇(分析纯,南京化学试剂股份有限公司);有机滤头(0.22 μm孔径,爱西默科技有限公司)。
次黄嘌呤(6-hydroxypurine,CAS:68-94-0,纯度≥98%),黄嘌呤(2,6-dihydroxypurine,CAS:69-89-6,纯度≥98%),以上对照品由上海源叶生物科技有限公司提供;硫酸-3-羟基喹啉(jineol-8-sulfate,纯度≥98%),硫酸-3-羟基-4-甲氧基喹啉(3-hydroxy-4-methoxyquinolin-8-yl hydrogen sulfate,纯度≥97%),3,8-二羟基喹啉(jineol,纯度≥98%),以上对照品为自制品。
1.2 仪器
Agilent 1100高效液相色谱仪(包括DAD检测器、四元低压梯度泵、AgilentOpen Lab色谱工作站,安捷伦科技有限公司);十万分之一电子分析天平(TOLEDO LE204E,METTLER);万分之一电子分析天平(01193-YP601N,METTLER);数控超声波清洗器(昆山洁力美超声仪器有限公司);纯水/超纯水系统。
1.3 试验材料
蜈蚣药材由鲁南厚普制药有限公司提供,蜈蚣药材共计14批,经鲁南制药集团高级工程师范建伟鉴定,均为蜈蚣科动物少棘巨蜈蚣ScolopendrasubspinipesmutilansL. Koch的干燥全体,凭证标本存放于鲁南制药集团中药制药共性技术国家重点实验室。样品具体信息如表1所示。
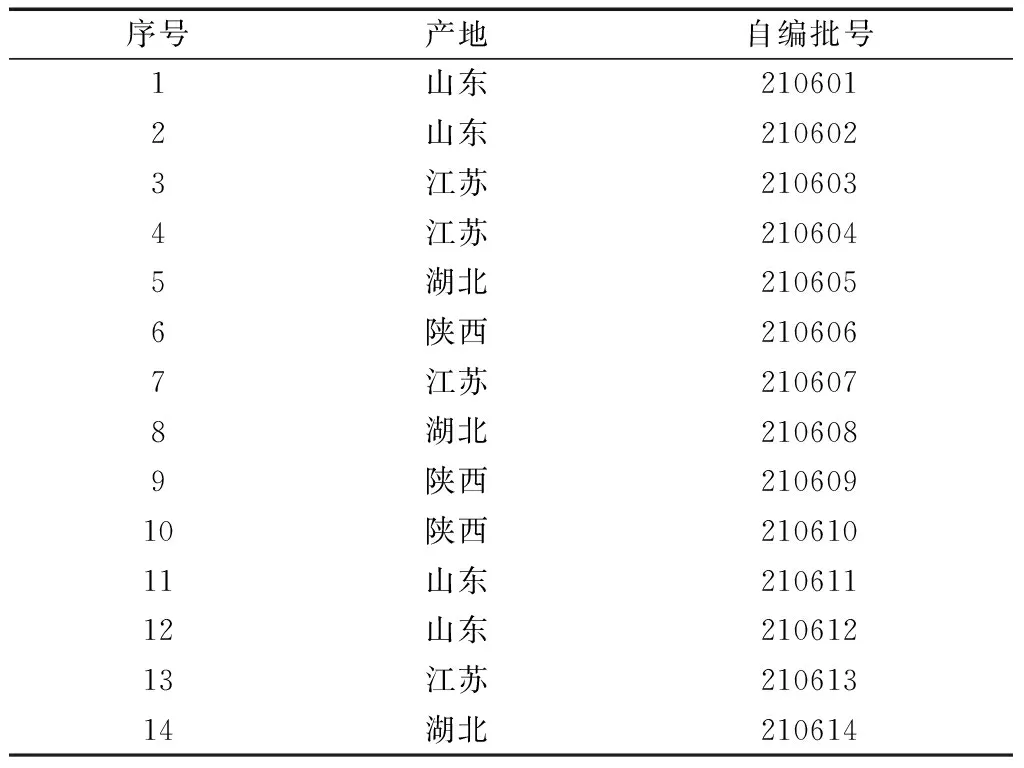
表1 样品信息表(n=14)
2 方法与结果
2.1 溶液制备
2.1.1 供试品溶液制备
取蜈蚣药材适量,粉碎机粉碎,过4号筛,作为供试样品,备用。取样品粉末2 g,精密称定,置于100 mL圆底烧瓶中,精密加入50%甲醇溶液50 mL,密塞,称重,回流30 min,放冷至室温,再称重,用50%甲醇溶液补足减失重量,摇匀,离心(4 500 r/min,5 min),吸取上清液,过0.22 μm有机滤头,弃去初滤液,以续滤液作为供试品溶液。
2.1.2 对照品溶液制备
精密称取次黄嘌呤、黄嘌呤、3,8-二羟基喹啉、硫酸-3-羟基喹啉、硫酸-3-羟基-4-甲氧基喹啉各适量,分别用纯甲醇溶解并制成浓度分别为0.131 1、0.381 0、0.331 6、0.274 8、0.298 0 mg/mL的对照品贮备溶液,备用。取上述对照品贮备液适量体积,稀释成次黄嘌呤、黄嘌呤、3,8-二羟基喹啉、硫酸-3-羟基喹啉、硫酸-3-羟基-4-甲氧基喹啉 0.065 55、0.038 1、0.041 45、0.010 992、0.029 8 mg/mL的溶液,即为混合对照品溶液。
2.2 色谱条件
色谱柱:YMC-Pack ODS-AQ(250 mm×4.6 mm,5 μm);流动相A:10 mmol/LKH2PO4溶液,流动相B:乙腈;流速:0.8 mL/min;溶剂:甲醇-超纯水(50∶50);柱温:30 ℃;进样量:5 μL;检测波长:254 nm;梯度洗脱(0~15 min,0%B;15~40 min,0%B变至20%B;40~60 min,20%B;60~65 min,20%B变至0%B),理论塔板数按次黄嘌呤计算不得低于5 000。蜈蚣药材高效液相色谱图如图1所示。
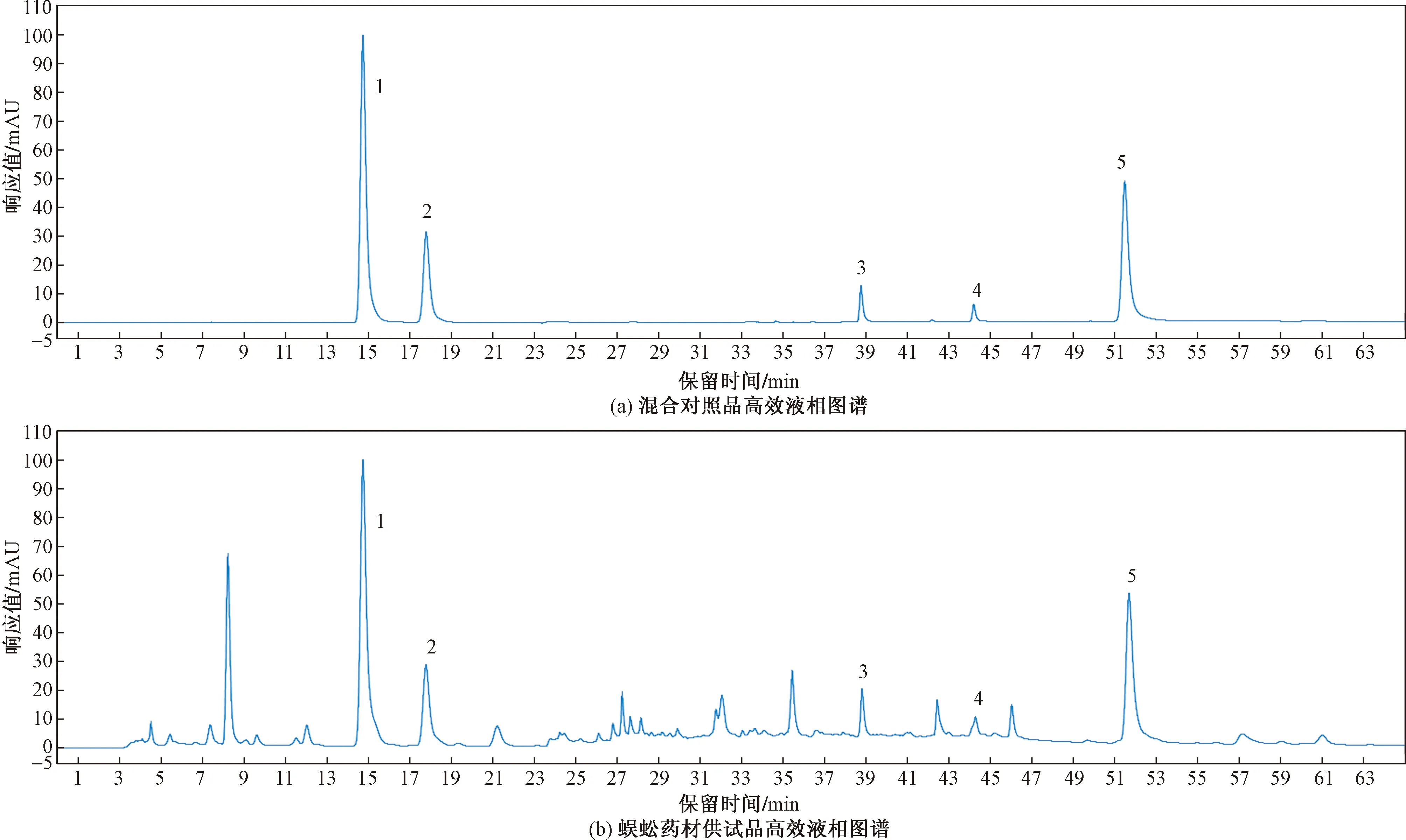
1为次黄嘌呤;2为黄嘌呤;3为硫酸-3-羟基喹啉;4为硫酸-3-羟基-4-甲氧基喹啉;5为3,8-二羟基喹啉图1 高效液相图谱Fig.1 HPLC chromatogram
2.3 方法学考察
2.3.1 线性范围考察
分别取上述混合对照品溶液不同体积,按照2.2节色谱方法进样,以进样浓度为横坐标(X),峰面积为纵坐标(Y),计算回归方程,绘制标准曲线,5种活性成分线性回归方程如表2所示。试验结果表明,次黄嘌呤、黄嘌呤、硫酸-3-羟基喹啉、硫酸-3-羟基-4-甲氧基喹啉、3,8-二羟基喹啉在各自相应的质量浓度范围内呈良好的线性关系。

表2 5种活性成分线性方程
2.3.2 精密度试验
取混合对照品溶液,按照2.2节色谱方法进样,连续进样6次,记录峰面积。得到次黄嘌呤、黄嘌呤、3,8-二羟基喹啉、硫酸-3-羟基喹啉、硫酸-3-羟基-4-甲氧基喹啉平均峰面积的相对标准偏差(relative standard deviation,RSD)见表3,5种成分的RSD均小于1.0%,表明本色谱方法系统适用,仪器精密稳定。
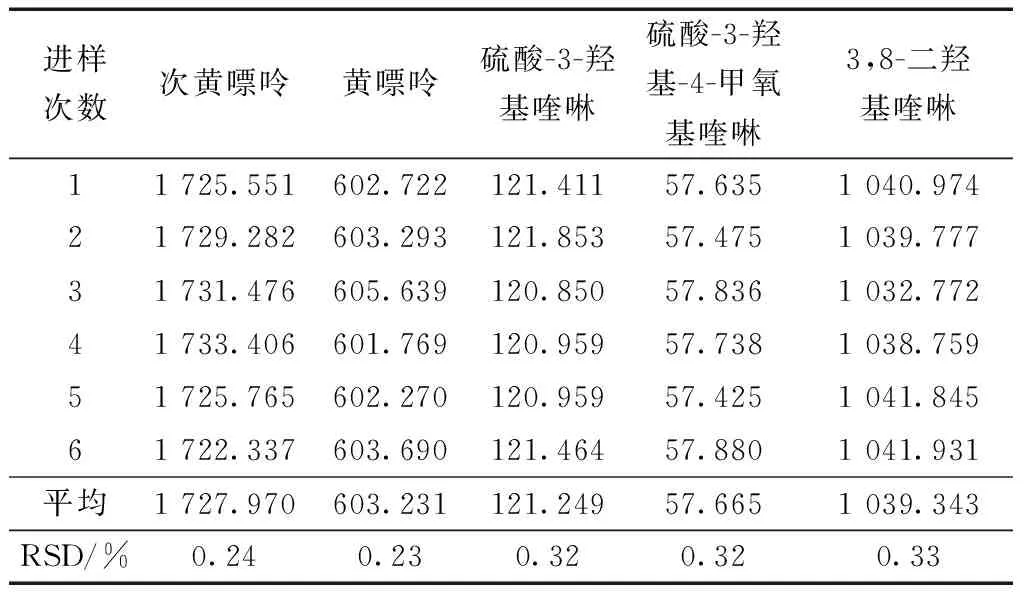
表3 5种活性成分精密度试验(n=6)
2.3.3 重复性试验
精密称取蜈蚣药材粉末6次,按照2.1节的供试品溶液制备方法制备供试品溶液,按照2.2节的色谱方法分别进样,记录峰面积,计算成分含量,结果见表4。各组分6次含量平均值的RSD均小于1.0%,表明蜈蚣药材活性成分含量测定方法重复性良好。
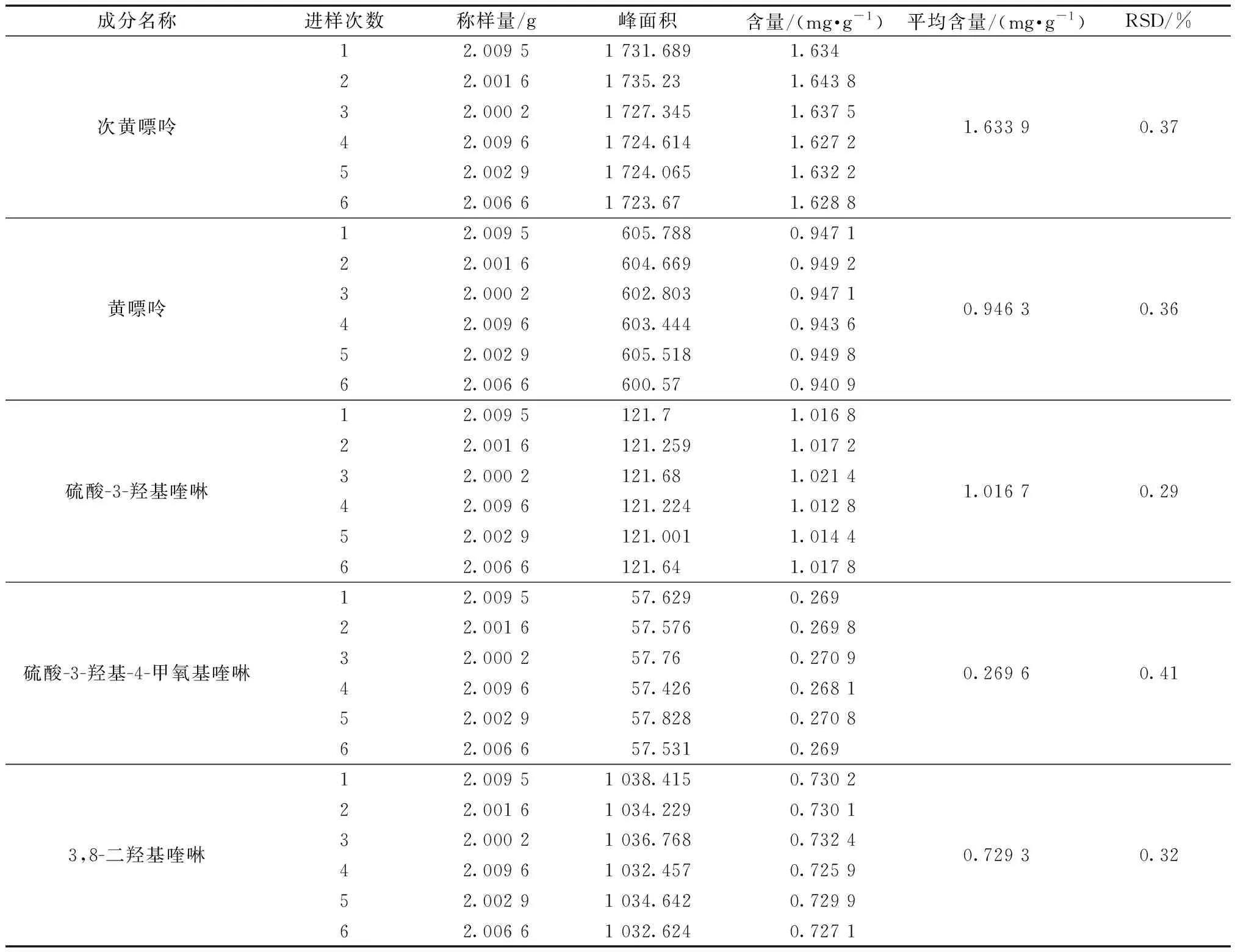
表4 5种活性成分重复性试验(n=6)
2.3.4 稳定性试验
精密称取蜈蚣药材供试样品,按照2.1节的供试品溶液制备方法制备供试品溶液,于0、2、4、6、8、12、24、48 h分别按照2.2节的色谱方法进样,记录峰面积。得到次黄嘌呤、黄嘌呤、3,8-二羟基喹啉、硫酸-3-羟基喹啉、硫酸-3-羟基-4-甲氧基喹啉平均峰面积并计算RSD,4种成分的RSD均小于2.0%,表明蜈蚣药材供试样品溶液在48 h内稳定,见表5。
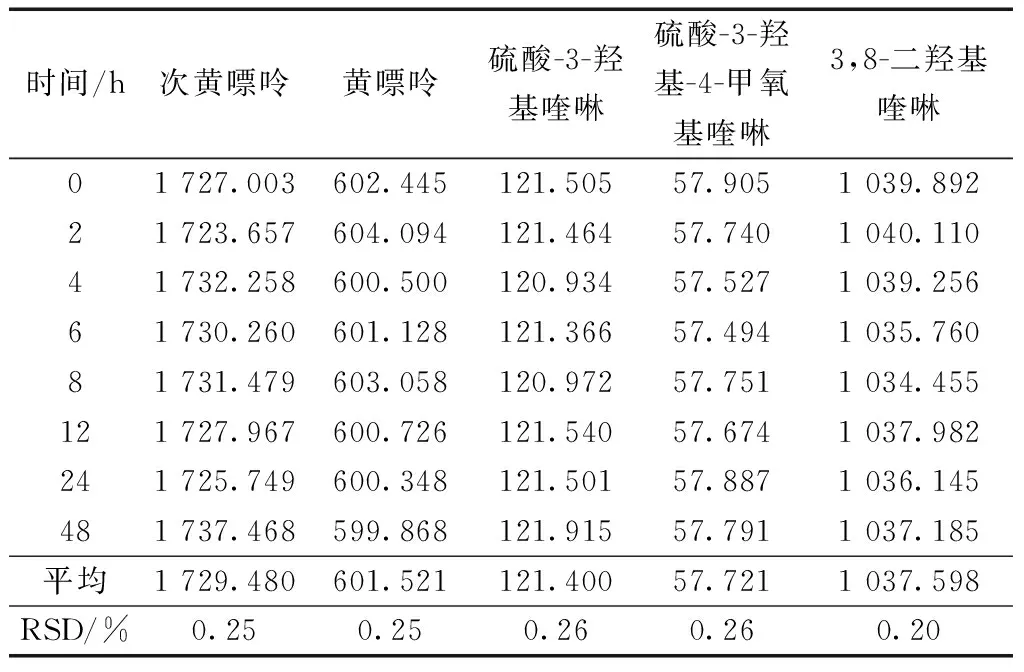
表5 5种活性成分稳定性试验
2.3.5 加样回收率试验
精密称取已知含量的蜈蚣药材样品粉末6份,分别准确加入含有等量各组分对照品的对照品,按2.1节的供试品溶液制备方法制备样品溶液,按照2.2节的色谱方法进样,所得结果如表6所示。结果显示,各组分平均回收率的RSD均小于4.0%,说明两个含量测定方法准确度良好。回收率的计算公式为
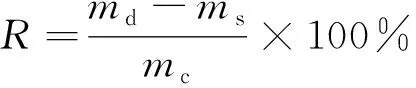
(1)
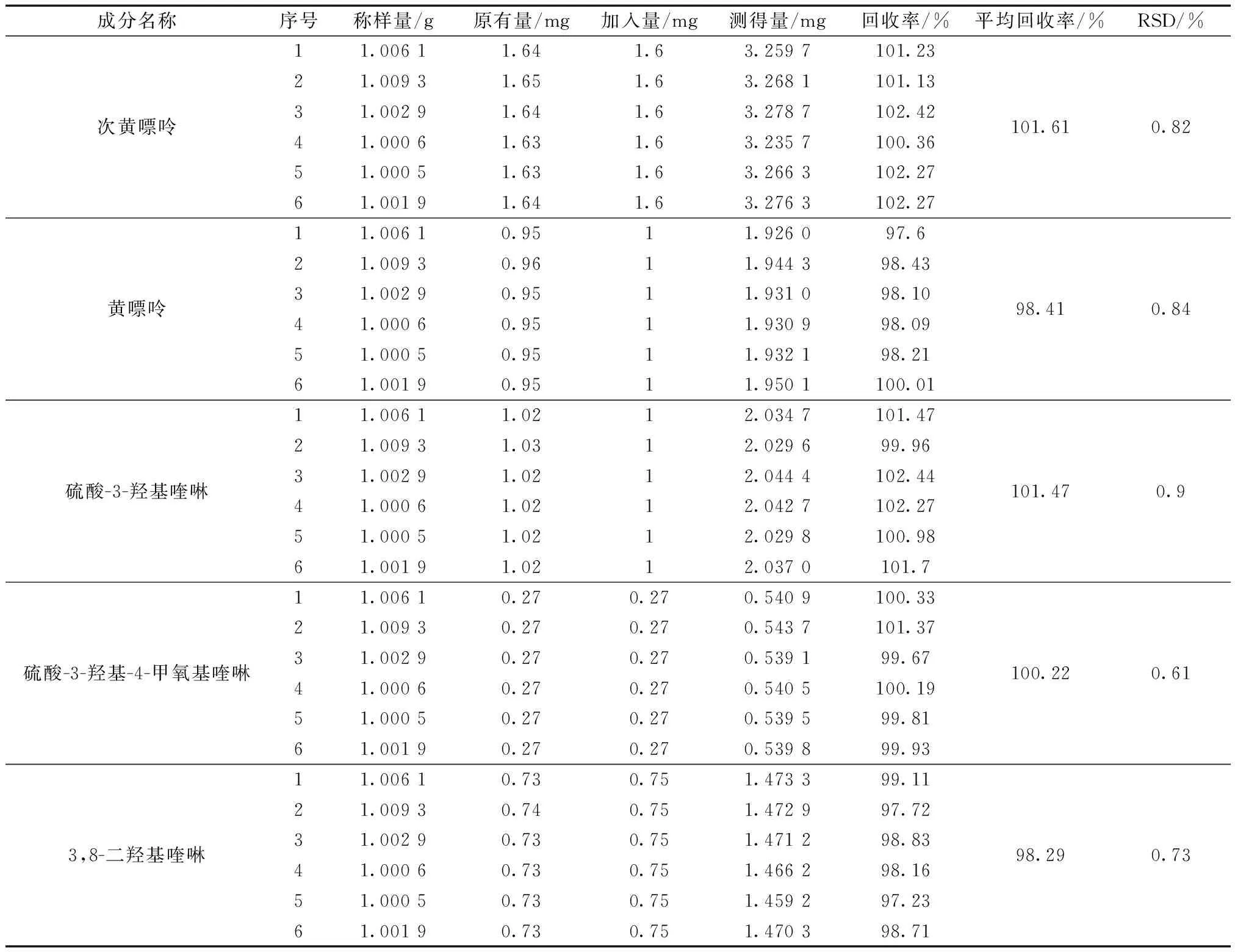
表6 5种活性成分回收率试验(n=6)
式(1)中:R为回收率;md为测得质量;ms为样品中所含被测成分质量;mc为加入的对照品质量。
2.4 一测多评法测定蜈蚣药材中5种成分含量
2.4.1 相对校正因子的计算
(1)斜率校正法。标准曲线表达式为
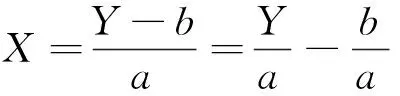
(2)
式(2)中:X为进样浓度;Y为峰面积;a为线性方程斜率;b为线性方程截距。
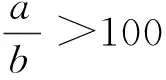
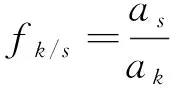
(3)
式(3)中:fk/s为校正因子;as为内参物s的线性方程斜率;ak为待测成分k线性方程斜率。
待测成分质量浓度计算公式为
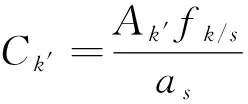
(4)
式(4)中:Ck′为待测成分浓度;Ak′为待测成分峰面积;fk/s为校正因子;as为内参物斜率。
应用斜率校正法进行计算,得到其他成分相对于次黄嘌呤的fk/s,黄嘌呤、3,8-二羟基喹啉、硫酸-3-羟基喹啉、硫酸-3-羟基-4-甲氧基喹啉相对于次黄嘌呤的fk/s见表7。
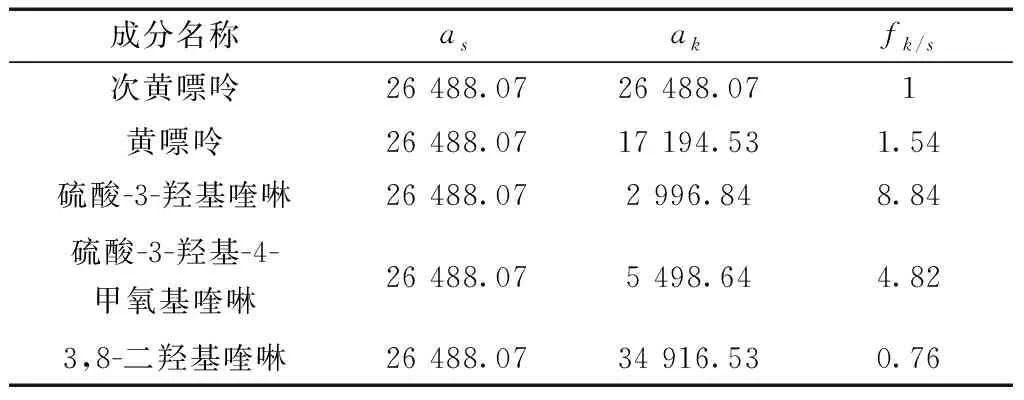
表7 斜率校正法校正因子
(2)多点校正法。以多个质量浓度点计算所得的fk/s取平均值做定量用fk/s。校正因子计算公式为
fk/s=(ASCK)/(AkCs)
(5)
待测成分质量浓度计算公式为
Ck′=(fk/sCsAk′)/AS
(6)
式中:AS为参照物质色谱峰峰面积;Cs为参照物质量浓度;AK为其他对照组分色谱峰峰面积;Ck为其他对照组分质量浓度;Ak为待测组分色谱峰峰面积。计算结果如表8所示。
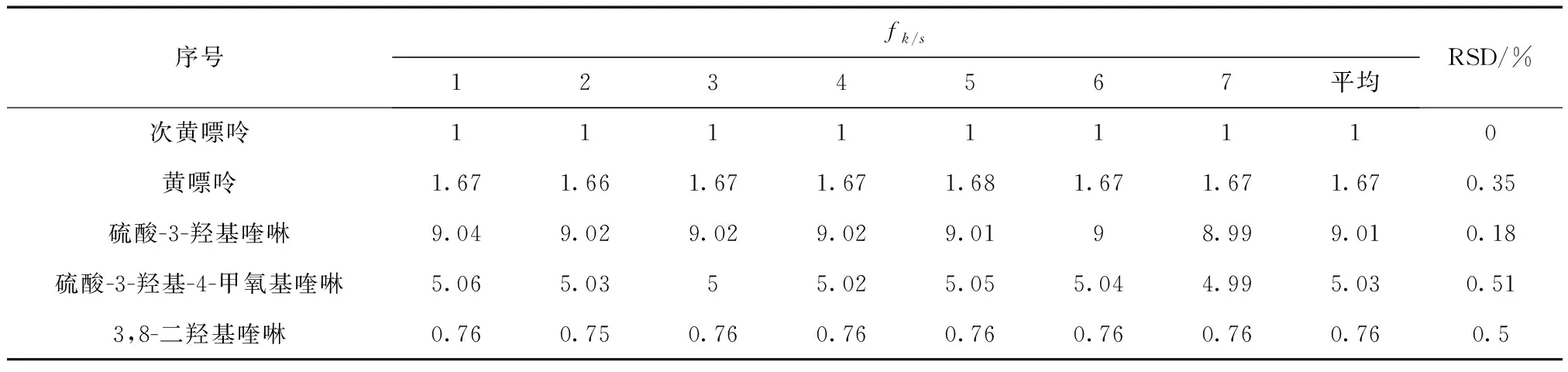
表8 多点校正法校正因子(n=5)
2.4.2 相对保留值的计算
利用相对保留时间来进行定位,即以各待测组分的色谱峰与次黄嘌呤色谱峰的保留时间来进行确定。相对保留时间计算公式为
RRT=Tk/Ts
(7)
式(7)中:Ts为参照物保留时间;Tk为其他待测组分保留时间。计算结果如表9所示。
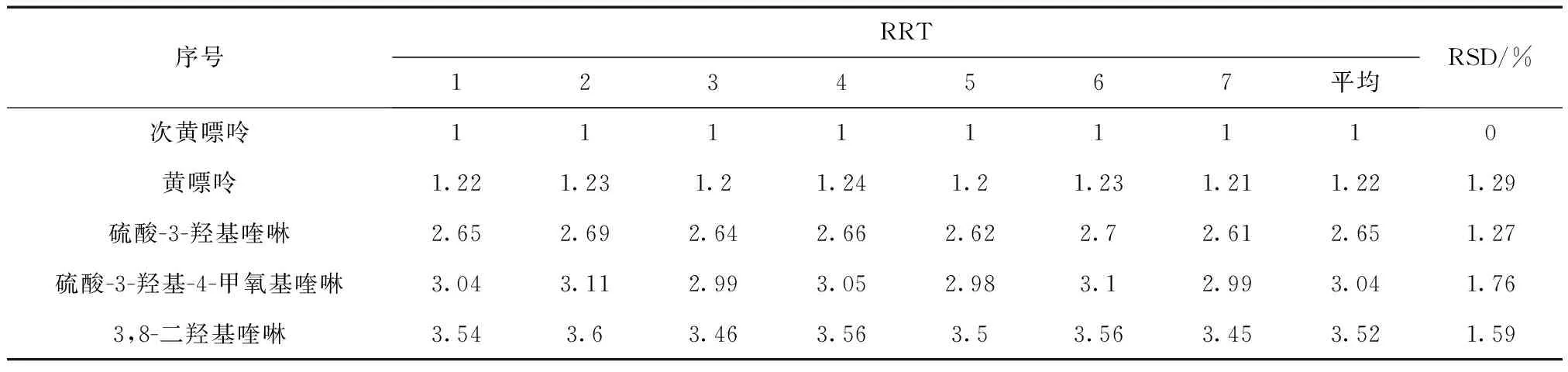
表9 多点校正法相对保留值(n=5)
2.5 一测多评方法在蜈蚣药材质量评价中的应用
2.5.1 一测多评结果与外标法含量测定结果比较
取14批蜈蚣药材供试样品按照2.1节的制备对照品溶液、供试品溶液,按2.2节的色谱条件进样,并分别通过一测多评方法与外标法对其进行含量测定,并比较两种测定方法的结果,见表10。由表10可知,不同计算方法得到的4种活性成分含量RSD均小于3%,适用性良好。
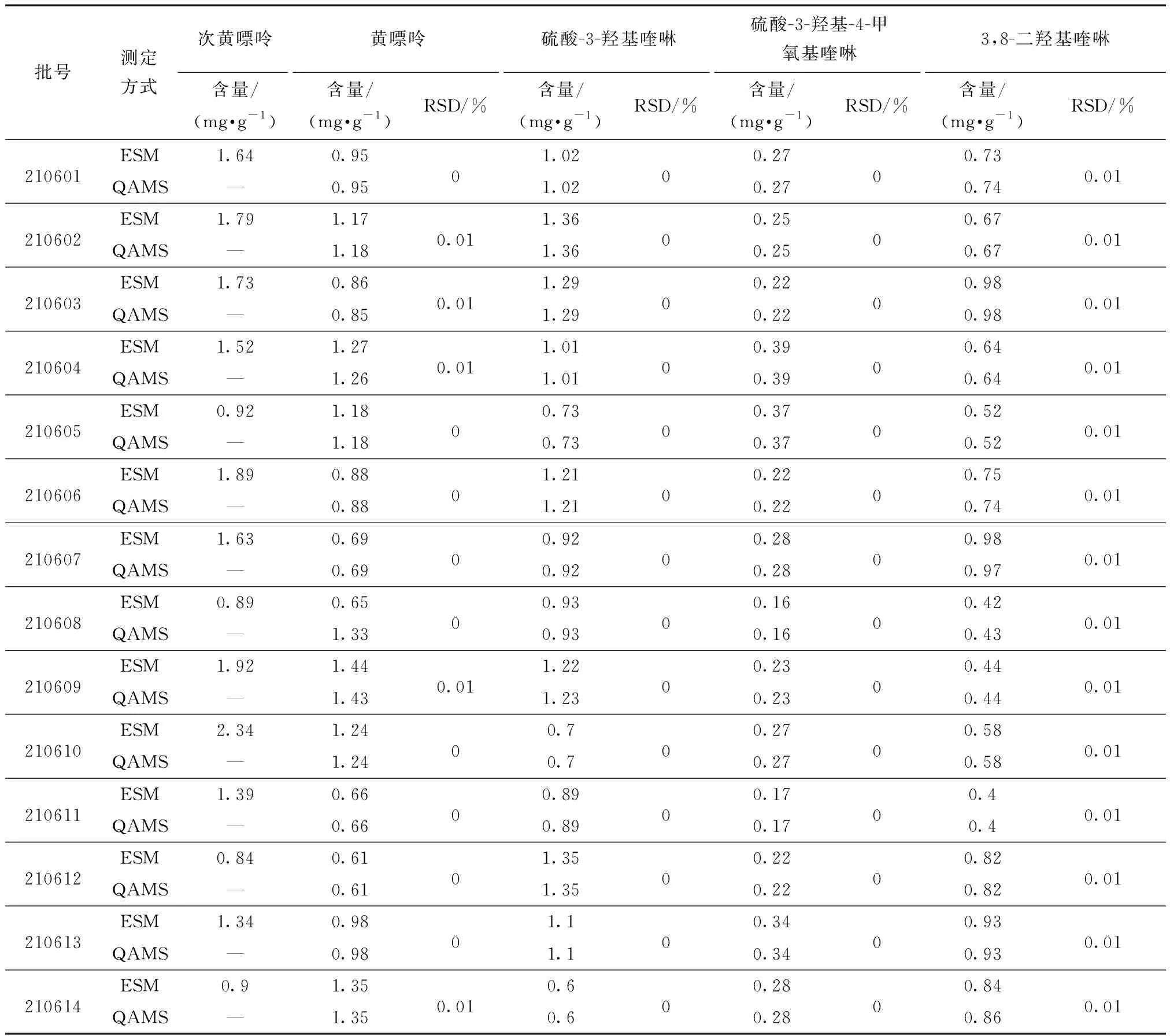
表10 一测多评结果与外标法含量测定结果(n=14)
2.5.2 多点校正相对保留值与外标实际保留时间比较
将14批蜈蚣药材供试品通过多点校正因子计算的理论保留时间与外标法的实际保留时间进行比较,结果无显著差异,见表11,5种活性成分计算保留值与实际保留值之间RSD均小于1.0%。
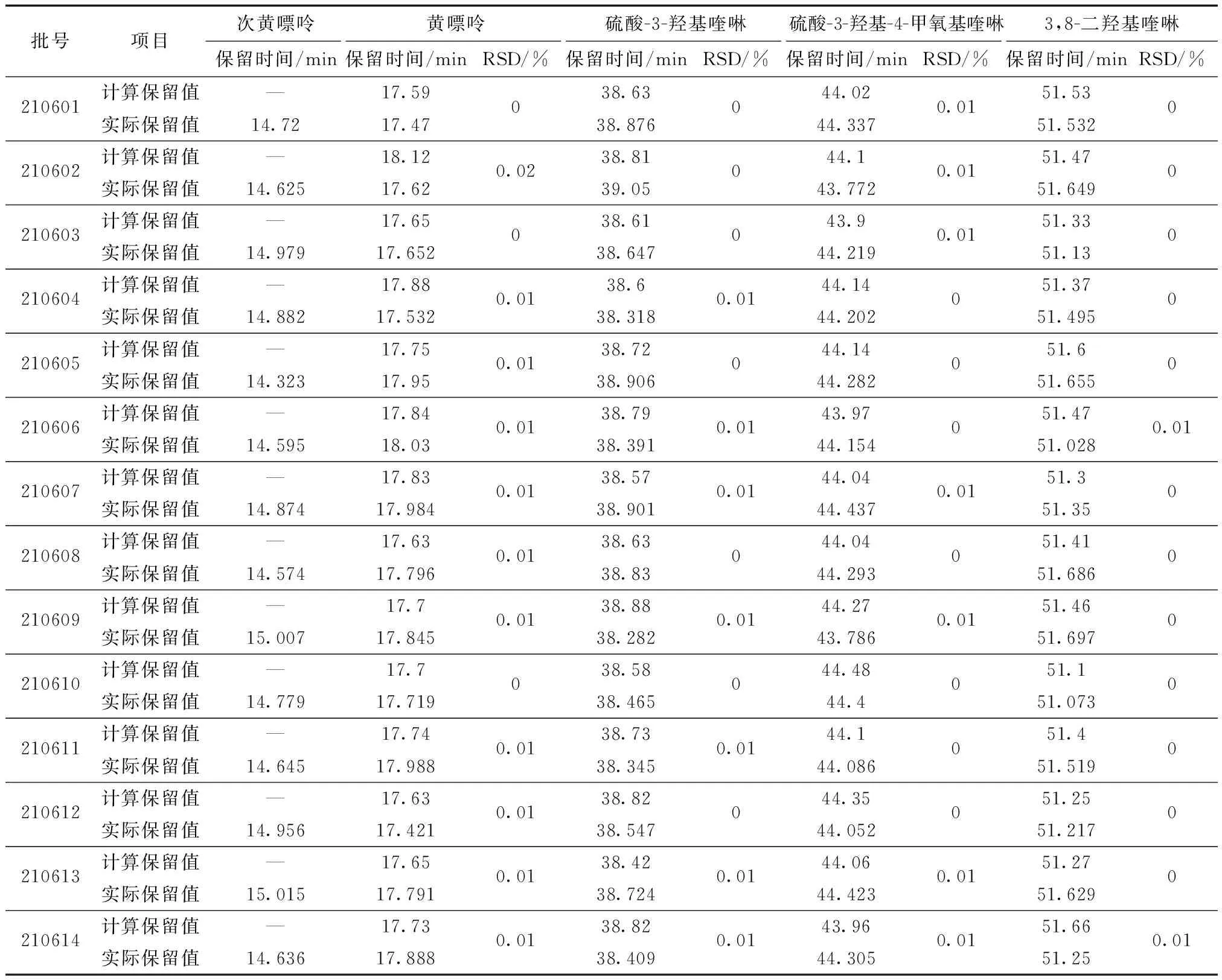
表11 一测多评结果与外标法保留时间比较(n=14)
3 讨论与结论
3.1 色谱条件确定
动物药材药效成分尚未清晰,蜈蚣药材中,次黄嘌呤出峰时间稳定,价值相对于其他成分较低,所以将次黄嘌呤确定为内参物。试验过程中,分别考察提取溶剂70%乙醇、50%甲醇、70%甲醇、纯甲醇等;称样量1.5、2.0、2.5 g;溶剂体积25、50、100 mL;回流处理时间20、30、40 min;当提取方法为:称样量2.0 g、溶剂体积50 mL、提取溶剂50%甲醇、回流处理时间30 min时,色谱图峰面积表现最好,所有优选出的提取方法为:精密称取2.0 g供试品粉末、加入50 mL50%甲醇溶液、回流处理30 min。测定方法共检测蜈蚣药材中5种活性成分的含量。通过对蜈蚣药材中5种活性成分的全波长扫描,对比5种成分的最大吸收波长,综合考虑,将色谱方法的检测波长确定为254 nm。对相同色谱条件不同柱温下出峰时间及含量测定准确度进行考察,分别考察了25、30、35 ℃,发现在30 ℃条件下,各色谱峰分离度达标,出峰时间适当。通过两种方法进行了校正因子的计算,分别为斜率校正法与多点校正法,斜率校正法仅通过各成分线性范围内的斜率进行计算,为将方程偏差考虑在内,综合实际,此方法计算得成分含量存在较大误差;而多点校正法为通过实际不同进样浓度的峰面积与浓度数值进行计算得到校正因子,在校正因子验证部分,发现此方法得到的校正因子计算结果与外标法无显著差异,因此选择多点校正法计算得到的校正因子。
3.2 结论
采用HPLC将多成分含量测定方法与一测多评相结合,建立的以次黄嘌呤为内参物一测多评定量分析方法用于蜈蚣药材的质量评价,蜈蚣药材内容物中4种活性成分相对于内参物的校正因子(fk/s)分别为1.67、9.01、5.03、0.76,相对保留值(RRT)分别为1.22、2.65、3.04、3.52。在14批蜈蚣药材供试品中,外标法与QAMS法含量测定结果比较,两者无显著差异,表明QAMS测定结果可靠。所建立的一测多评方法具有稳定、准确、简单、快捷、成本低等特点,较全面地反映蜈蚣药材的内在质量,检测成本和检测时间均大幅降低,适用于蜈蚣药材优质标准的建立,有利于进一步加强动物药材的质量控制水平。
猜你喜欢
现代食品(2022年3期)2022-03-24
应用化工(2020年6期)2020-07-30
理化检验-化学分册(2020年4期)2020-06-03
启蒙(3-7岁)(2019年5期)2019-06-27
中成药(2017年7期)2017-11-22
小学生导刊(2017年14期)2017-05-17
山东农业科学(2017年2期)2017-03-15
河南科技学院学报(自然科学版)(2016年3期)2016-03-30
中国与非洲(法文版)(2016年8期)2016-02-11
红领巾·萌芽(2015年9期)2015-09-10
