
尼拉帕尼合成研究进展(二)
2023-02-02 06:55陈慧杰王秀军
精细石油化工 2023年1期
陈慧杰,王秀军
(江苏海洋大学药学院,江苏 连云港 222005)
(上接2022年第6期)
4 以丁二酸酐(21)为起始原料
2014年,Chung等[6]报道了以丁二酸酐(21)为起始物料的合成方法:在三氯化铝催化下,琥珀酸酐与溴苯发生傅-克酰基化反应生成酮酸化合物22,(22)经Fischer酯化和环氧化/重排生成芳香醛酯化合物25,而后在甲苯溶液中转化为亚硫酸氢盐化合物26,(26)在ATA-302酶催化下不对称合成化合物27,再经硼氢化钠还原、对甲苯磺酸成盐的关键中间体30,而后(30)与化合物31进行C—N偶联反应、脱保护和成盐反应得尼拉帕尼对甲苯磺酸盐,合成路线总收率为40%(见图5)。
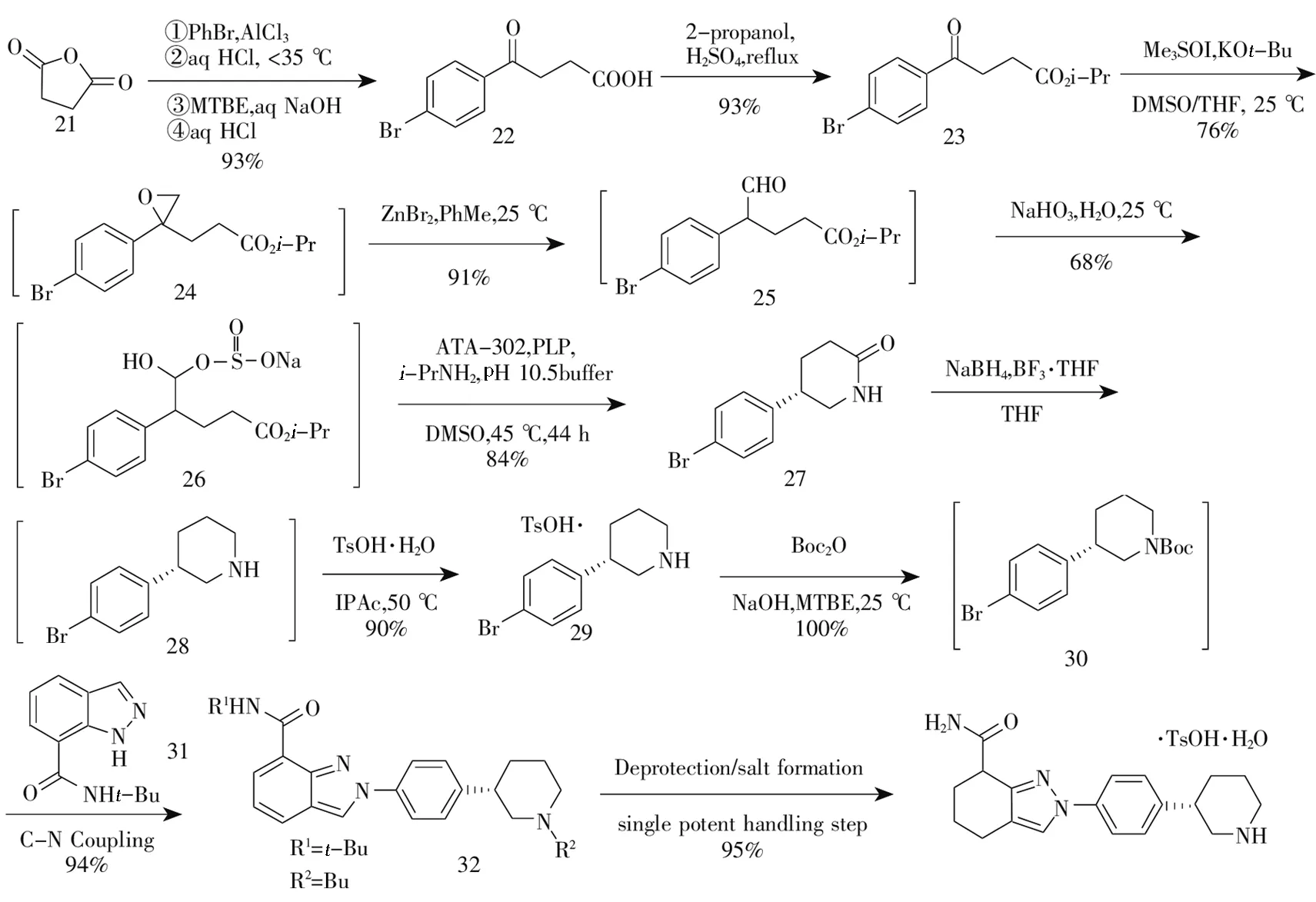
图5 尼拉帕尼合成路线四
该路线首次采用酶催化定向合成手性化合物,解决了手性分离困难的问题;另外,该路线充分利用吲唑环上取代基的空间位阻效应达到N2位定向C—N偶联反应,该反应具有较高的化学和区域选择性。
5 以4-溴苯基乙酸(33)和1-溴-3-氯丙烷(34)为起始原料
Chung等[6]以4-溴苯乙酸和1-溴-3-氯丙烷为起始原料,经缩合反应得化合物35,然后在乙酸异丙酯中经DBU催化合成内酯化合物36,(36)被还原后得到半缩醛(37),半缩醛在ATA-301酶催化下得单一旋光异构体(38),化合物38氨基经保护后与甲烷磺酰氯反应、分子内环化得到关键中间体30,而后通过C—N偶联反应、脱保护和成盐反应得尼拉帕尼对甲苯磺酸盐,合成路线总收率28%(图6)。路线五提供了另一种酶催化方法,为手性催化合成提供一条很好的思路。

图6 尼拉帕尼合成路线五
6 以苯基溴化镁(42)和N-苄基-3-哌啶酮(41)为起始原料
2018年,崔效源等[7]报道了以N-苄基-3-哌啶酮和苯基溴化镁为起始原料的合成方法:通过格氏反应得到3-羟基-3-苯基-1-苄基哌啶盐酸盐(43),(43)经消除反应和催化氢化反应得外消旋化合物3-苯基哌啶(45),通过手性拆分、硝化和还原反应得到了关键中间体(S)-4-(3-哌啶基)苯胺(48),中间体(48)经氨基保护与芳香醛5发生缩合反应形成席夫碱化合物50,而后经叠氮化、环化反应和酰胺化反应得尼拉帕尼保护产物17,化合物17经水解后通过酒石酸盐拆分和成盐反应得尼拉帕尼对甲苯磺酸盐,工艺总收率为4. 9%,终产品纯度为 99.9%(HPLC),ee值为99.9%,见图7。该工艺首次发现以N-苄基-3-哌啶酮和苯基溴化镁格氏试剂为起始原料合成3-苯基哌啶的新方法,可有效降低采用三吡啶硼酸和对碘硝基苯生产的成本。
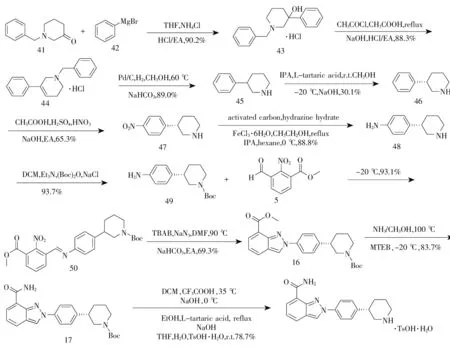
图7 尼拉帕尼合成路线六
7 以2-溴3-甲酰基-苯甲酸甲酯(51),S型叔丁基-3-(4-氨基苯基)哌啶-1-羧酸叔丁基酯(49)和四丁基叠氮化铵(TBAA)(52)为起始原料
2018年,吴学平等[8]报道了以2-溴3-甲酰基-苯甲酸甲酯(51)、S型叔丁基-3-(4-氨基苯基)哌啶-1-羧酸叔丁基酯(49)和四丁基叠氮化铵(52)为原料的微通道反应方法。该专利合成尼拉帕尼的路线分3步:首先将化合物51,化合物49和TBAA分别配置成溶液,通过注射器加入到微通道反应器中,经微通道反应合成化合物16,而后,化合物16在DMF溶剂中,经甲醇钠和甲酰胺催化氨解得到化合物17,最后经三氟乙酸脱保基得尼拉帕尼,见图8。该法首次采用微通道反应合成关键中间体16,极大地提高了反应效率,有利于大规模工业化生产。
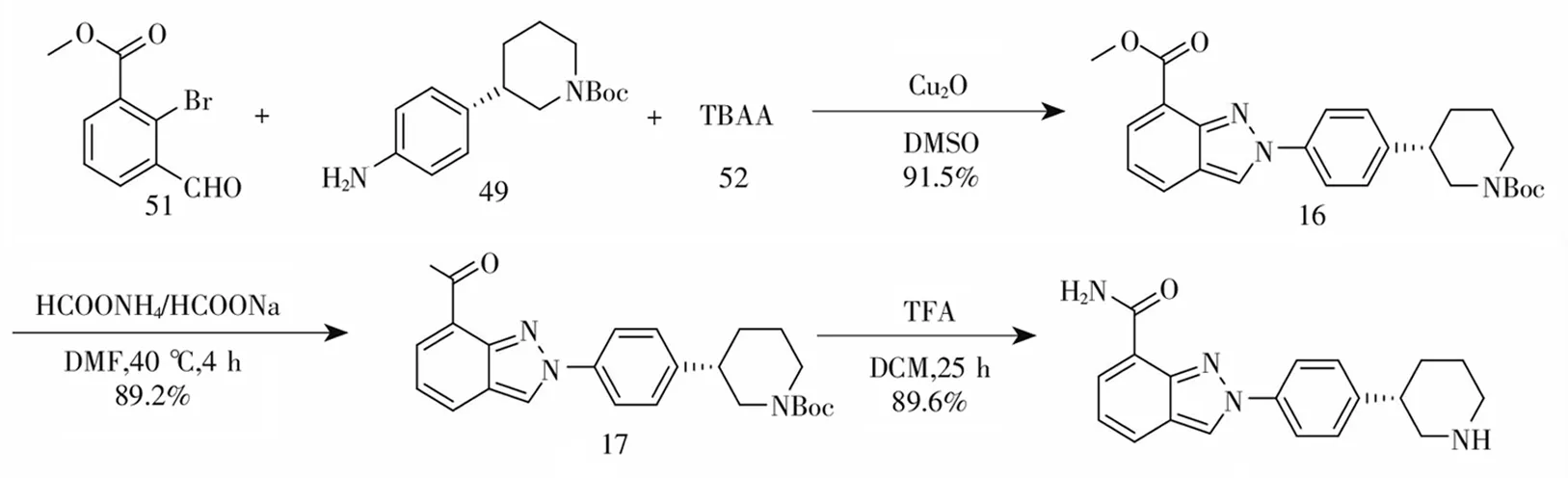
图8 尼拉帕尼合成路线七
8 以2-氨基苯甲酰胺(53)和(S)-4-(吡啶-3-基)苯胺(48)起始原料
2019年,刘长春等[9]以化合物53和中间体48为起始原料,以溴化亚铜为催化剂,吡啶为配体,叔丁基过氧化氢(TBHP)为氧化剂,采用微波加热脱氢偶联得化合物54,而后在二氯(对甲基异丙基苯基)钌二聚体和乙酰丙酮钴催化下,与多聚甲醛经微波加热环化得到尼拉帕尼(图9)。该工艺两步反应均采用微波加热,微波加热反应条件温和,时间较短,但工业化放大不易实现。
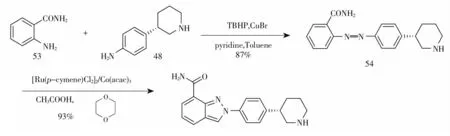
图9 尼拉帕尼合成路线八
9 以3-溴吡啶(56)与苯硼酸酯(55)为起始原料
2017年,王雪根等[10]开发了以3-溴吡啶与苯硼酸酯为原料合成尼拉帕尼关键中间体30的方法,两种原料通过Suzuki偶联反应合成化合物57,而后经溴代得化合物58,化合物58在铑碳催化下通入氢气得化合物59,再经手行拆分和氨基保护得到关键中间体30,中间体30与化合物60进行Ulman反应后得化合物16,化合物16再经氨解和成盐得到尼拉帕尼对甲苯磺酸盐(图10)。该路线降低了路线四的生产成本。

图10 尼拉帕尼合成路线九
10 以2-碘苯基-1,3-二(三氟甲烷磺酸酯)(61)为起始原料
2017年,Shi等[11]以2-碘苯基-1,3-二(三氟甲烷磺酸酯)为起始原料,在苄基溴化镁作用下形成3-三氟氧基苯炔中间体,与化合物62环加成反应得到化合物63。而后化合物63与化合物64继续环加成反应得到化合物65,最后经过脱保护基和氨基得到尼拉帕尼,总收率为14.2%。化合物64可由中间体3-(4-氨基苯基)哌啶-1-羧酸叔丁酯(19)制得,见图11。该路线较为新颖,但原料来源不易,第二步环加成反应会产生大量异构体2H-吲唑-4-乙酸甲酯(65′),导致总产率不高,不适合工业大生产。
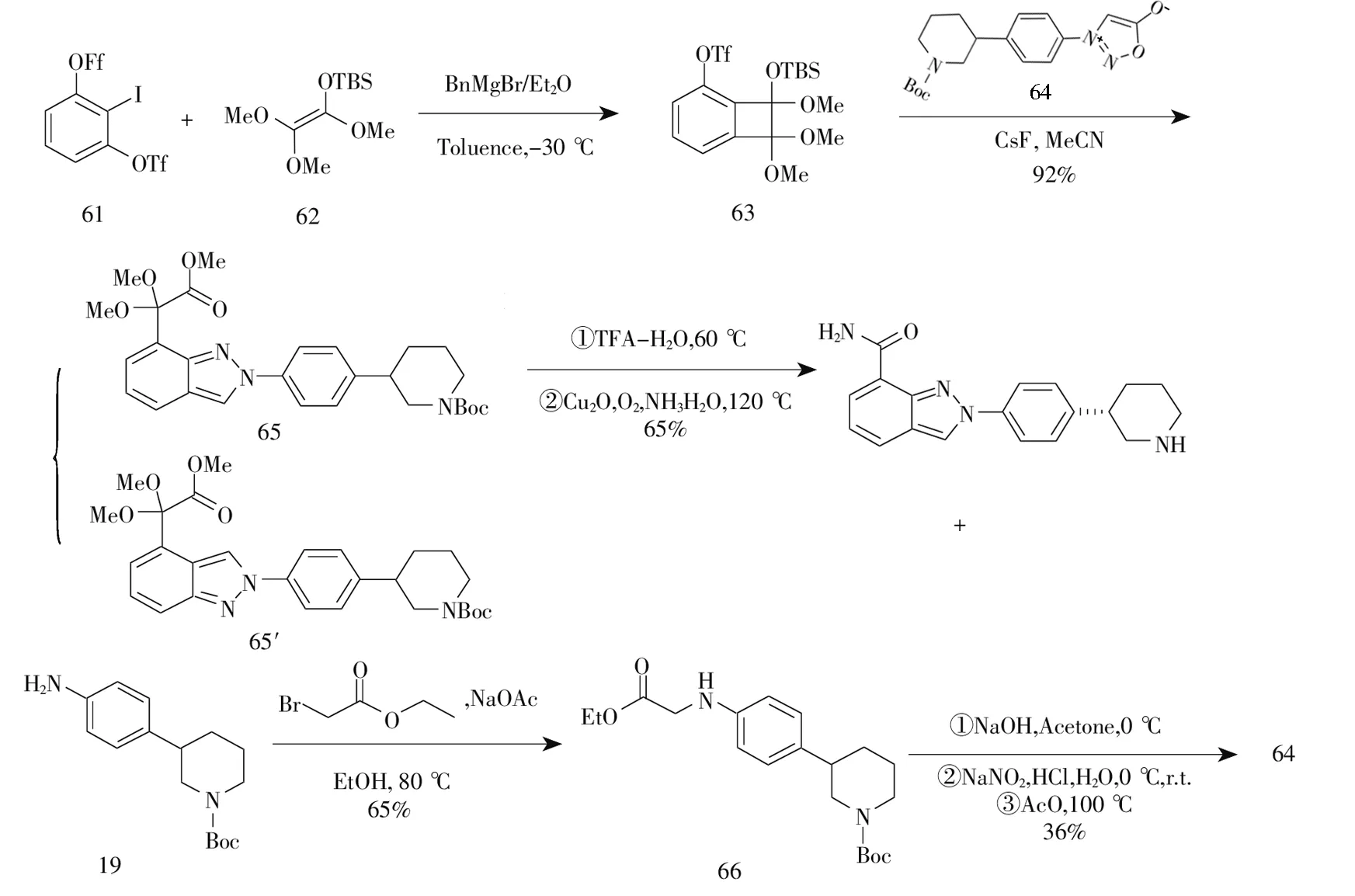
图11 尼拉帕尼合成路线十
11 结束语
综上所述,现有尼拉帕尼的合成方法大都包含S型叔丁基-3-(4-氨基苯基)哌啶-1-羧酸叔丁基酯的合成、吲唑环的合环和Ulman反应,合成路线各有利弊。
原研专利路线收率偏低,使用了叠氮化钠,安全系数很低,且采用手性SFC进行拆分。Wallace等开发了尼拉帕尼公斤级生产的合成路线,但反应过程需用到酒石酸盐拆分,并且采用叠氮化钠合环,安全系数和收率较低。路线六以N-苄基-3-哌啶酮和苯基溴化镁格氏试剂为起始原料合成3-苯基哌啶,可降低关键中间体S型叔丁基-3-(4-氨基苯基)哌啶-1-羧酸叔丁基酯的生产成本;路线七采用微通道反应合成尼拉帕尼可将两步反应的收率提高至80.9%;路线八采用微波反应和多聚甲醛反应替代叠氮化钠进行合成,虽降低了安全风险,提高了反应效率,但工业化生产不易实现;路线九以3-溴吡啶与苯硼酸酯为原料合成尼拉帕尼关键中间体S型叔丁基-3-(4-溴苯基)哌啶-1-羧酸叔丁基酯,该路线可降低生产成本;路线十的合成方法十分新颖,但是原料来源不易,反应过程中产出副产物较多,工业化生产较难实现。Chung等首次提出了酶催化合成关键中间体(S)-5-(4-溴苯基)哌啶-2-酮的方法,极大的提高了反应立体选择性;另一方面,吲唑2位N原子直接和S型叔丁基-3-(4-溴苯基)哌啶-1-羧酸叔丁基酯合成极大的提高了产品的收率,该工艺具有很大的工业化市场前景。随着酶催化和合成技术的发展,将会出现更多成本低、收率高和更加安全环保的尼拉帕尼合成工艺。(续完)
猜你喜欢
健康体检与管理(2022年2期)2022-04-15
化学与生物工程(2021年8期)2021-08-26
法人(2021年12期)2021-05-09
国外医药(抗生素分册)(2016年2期)2016-07-12
合成化学(2015年1期)2016-01-17
应用化工(2014年1期)2014-08-16
应用化工(2014年3期)2014-08-16
中国药理学通报(2014年2期)2014-05-09
郑州大学学报(理学版)(2014年4期)2014-03-01
应用技术学报(2014年1期)2014-02-28
