
基于分子对接、分子动力学模拟虚拟筛选中药来源的醛脱氢酶1A1 抑制剂
2023-01-31 08:43:24吴辉渊邓兰李芬芬
中国现代药物应用 2022年24期
吴辉渊 邓兰 李芬芬
治疗耐药性是临床肿瘤学面临的最大挑战之一[1],恶性肿瘤复发源于耐药性肿瘤细胞能够重新填充肿瘤生态位并播种新的病变,这些细胞被称为癌症干细胞(cancer stem cells,CSCs),CSCs 具有肿瘤原始特性、干细胞性和侵袭性,其重要特征之一是高醛脱氢酶1A1(aldehyde dehydrogenas 1A1,ALDH1A1) 表达[2]。ALDH1A1 主要存在于肿瘤干细胞表面及胞浆,作为CSCs 标志物,表达水平越高患者预后越差,与ALDH1A1 相关的抑制CSCs 增殖和耐药的抗癌治疗逐渐受到重视,目前尚无以ALDH1A1 为靶点的药物上市[3]。NCT-501 对 ALDH1A1 具有较高的选择性抑制作用,是目前ALDH1A1 活性研究中常用的阳性对照药[4]。中药活性成分以种类多样、结构独特、生物活性高等特点,在抗肿瘤领域具有突出的优势。本研究利用类药性筛选,分子对接,分子动力学模拟等一系列技术,从中药化合物库筛选出潜在的ALDH1A1 抑制剂活性物质,提供后续临床及药物研究的基础。
1 材料与方法
1.1 材料 本研究采用Ledock 软件进行分子对接工作。ADME 预测采用SwissADME 在线工具,分子动力学模拟使用AMBER 18 软件进行。
实验仪器与试剂:ALDH1A1 重组蛋白购于上海泽叶生物科技有限公司,血竭素(Cochinchinenin)购于成都植标化纯生物技术有限公司;NCT-501 购于上海脉铂医药科技有限公司,酶标仪为Spark 10 M 型,氧化型辅酶1(NAD),二甲基亚砜、乙醛均购于美国sigma,其他试剂均为国药分析纯。
1.2 靶蛋白ALDH1A1 结构预处理 虚拟筛选使用的ALDH1A1 蛋白晶体结构从PDB 数据库[5]中下载获得,PDB ID 为4WPN;在对接开始之前,使用对接软件中的Lepro 模块对受体蛋白进行处理,包括去除水分子、盐离子以及小分子等操作。准备靶标ALDH1A1蛋白活性口袋并产生格点文件,原结晶配体坐标作为格点盒子中心。4WPN 晶体结构中蛋白与小分子抑制剂CM053 结合模式及关键残基见图1。
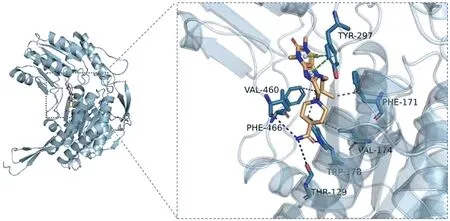
图1 4WPN 结合模式及关键残基
1.3 中药分子化学结构的预处理 配体小分子库含17580 个常见的天然产物分子,该数据库从陶术生物官方(https://www.tsbiochem.com/natural-products)获取。使用Openbabel 2.4.1 对其进行2D 至3D 格式转换工作。
1.4 类药性评估 基于类药性规则“Lipinski Ro5”对中药分子配体库进行首轮筛选,筛选出10965 个符合类药性规则的命中分子。Lipinski Ro5 筛选规则[6]:分子量(MW):500~700 Da,化合物的油水分配系数的对数值(clog P)=0~7.5,氢键供体(HBD)≤5,氢键受体(HBA)≤10,拓扑极性表面积(TPSA)≤200 Å2,可旋转键的数量(NRB)≤20。
1.5 分子对接 设置对接盒子,使用PyMol 插件center_of_mass.py 基于活性位点的位置定义对接盒子的中心(37.33,16.60,14.42),盒子边长设定为18 Å。设定每个分子对接输出最大构象数为3 个,其余参数保持默认设置。输出的打分最高的对接构象被我们认为是结合构象,选取Score<-10 kcal/mol 分子。
1.6 ADME 预测 基于ADME 药动学参数,采用SwissADME 在线工具对Score<-10 kcal/mol 分子进行ADME 预测。从MW、HBD、HBA、脂水分配系数(lipid-water partition coefficient)、可旋转键(rotatable bonds)、生物利用度(bioavailability)、TPSA、脑血管障壁(blood-Brain Barrier,BBB)、P-糖蛋白(P-gp)、水溶性ESOL Log S 等评估,筛选具有良好类药性质的化合物。
1.7 分子动力学模拟 基于上述对接获得的小分子与蛋白复合物作为初始结构分别进行全原子分子动力学模拟,模拟使用AMBER 18 软件进行。模拟之前,用antechamber 模块和gaussian 09 软件的 HF/6-31G*计算中药分子的电荷。之后,中药分子采用GAFF2 力场[7],ALDH1A1 采用蛋白力场ff14SB[8]。各体系均采用LEaP 模块给体系添加氢原子,在体系10 Å 距离处添加截断的八面体TIP3P 溶剂盒,并在体系中添加Na+/Cl-用于平衡体系电荷,最后输出用于模拟的拓扑和参数文件。
分子动力学模拟采用AMBER 18 软件。在模拟之前,对体系进行能量优化,包括2500 步的共轭梯度法和2500 步的最速下降法。能量最小化后,在恒定升温速度和固定体积条件下,在200 ps 内使体系温度缓慢加热到298.15 K。维持298.15 K 的体系温度,500 ps的nvt 系综的平衡模拟,使溶剂盒子中的溶剂分子更均匀分布。然后,对平衡后的体系在NPT 条件下模拟500 ps。最后在周期边界性条件下分别对两个复合体系进行100 ns 的NPT 系综模拟。模拟时,设置10 Å 为非键的截断距离,运用Particle mesh Ewald 方法计算长程的静电作用[9],限制氢原子键长的方法选用SHAKE,温控用Langevin 算法[10],其中积分步长为2 fs,体系压强为1 atm(1 atm=1.01×105Pa),碰撞频率γ 设为2 ps-1,每10 ps 保存轨迹数据用于进一步分析。
1.8 MM/GBSA 结合自由能计算 所有体系的蛋白和配体间的结合自由能通过MM/GBSA 方法计算[11-13]。本研究中采用45~50 ns 的分子动力学模拟轨迹用作计算,具体公式如下。

其中,△Einternal表示内能、△EVDW表示范德华作用以及△Eelec表示静电相互作用。内能包括扭转能(Etorsion)、角能(Eangle)、和键能(Ebond),△GGB和△GGA统称溶剂化自由能。其中,GGB 为极性溶剂化自由能、GSA 为非极性溶剂化自由能。△GGB采用Nguyen[14]研究的GB 模型计算(igb=2)。△GSA 则采用表面张力(γ)与溶剂可及性表面积(surface area,SA)相乘得到,△GSA=0.0072×△SASA。
1.9 酶促反应动力学实验 乙醛脱氢酶1 的测试方法:通过测定波长340 nm 处吸光度值每分钟的变化,可计算出每分钟NAD+转化为还原型辅酶1(NADH)的量,从而得到该酶的酶活。定义每分钟波长340 nm 处吸光度值上升1 为1 个活力单位(U)。将5 μl 浓度为150 nM ALDH1a1 加入测定孔中,反应体系在25℃,包括1 mM EDTA,2 mM NAD+,0.1 M 磷酸缓冲溶液(pH=7.4)加入测定孔,最后加入250 μM 乙醛开始反应,并开始记录光密度(OD)值的变化,总体系反应时间为5 min,反应体系为300 μl。酶活性为△OD/min,重复试验组3 次。
2 结果
2.1 虚拟筛选流程及结果 对接模拟技术是探究小分子与目标靶点相互作用的便捷有效手段。基于此,本研究针对ALDH1A1 蛋白的活性位点,使用ledock 软件对常见的天然产物分子进行基于对接的虚拟筛选研究。中药分子库中共有17580 个中药单体化合物,进行多轮虚拟筛选,见图2。通过分子对接,筛选出排名前10 的命中分子,见表1。进一步ADME 预测,见表2。根据MM/GBSA 结合自由能,选取排名前4 的分子进一步做分子动力学模拟。见图3。四个中药化合物的名称为血竭素(Cochinchinenin)、波棱酮(Herpetone)、2,3 二氢橡胶树双黄酮(2,3-Dihydroheveaflavone)、苏铁双黄酮(Sotetsuflavone),对接软件对以上化合物的结合能打分分别为-10.692、-10.309、-10.359、-10.611 kcal/mol。
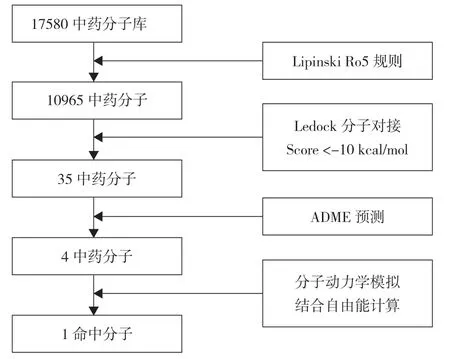
图2 虚拟筛选流程
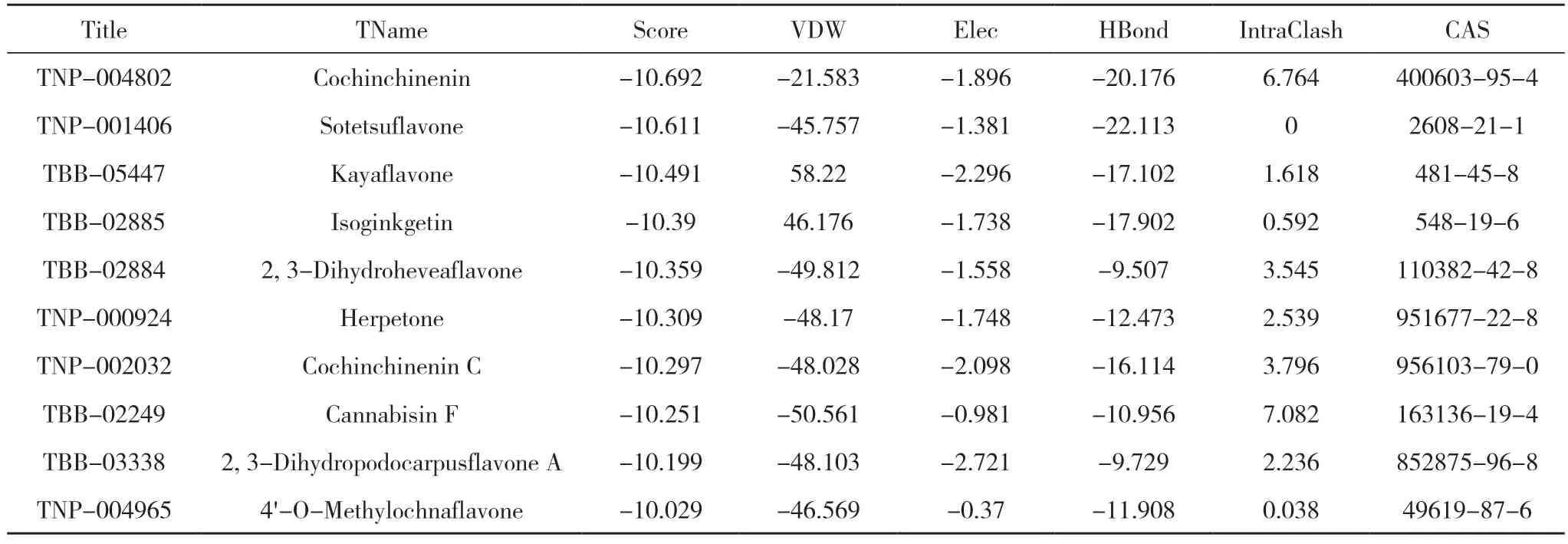
表1 分子对接(排名前10)

表2 ADME 参数预测(排名前4)

图3 基于虚拟筛选获得的4 个潜在的ALDH1A1 抑制分子
2.2 结合模式分析 基于上述虚拟筛选对接输出的Cochinchinenin、Herpetone、2,3-Dihydroheveaflavone、Sotetsuflavone 与ALDH1A1 蛋白的复合物进行结合模式分析。见图4。小分子2,3-Dihydroheveaflavone、Sotetsuflavone、Cochinchinenin、Herpetone 与ALDH1A1蛋白的结合模式:小分子2,3-Dihydroheveaflavone 与蛋白上的SER-461、TRP-178、THR-129、PHE-171、HIS-293 形成氢键作用,还和蛋白上的PHE-171 形成疏水作用。见图4A。小分子Sotetsuflavone 和蛋白上的SER-461、THR-129、TRP-178、TRP-297、HIS-293形成氢键作用,和LYS-128、VAL-174、ILE-304 形成疏水作用。此外,Sotetsuflavone 和ALDH1A1 蛋白上的TYR-297、PHE-171 还形成pipi-共轭作用。见图4B。对于Cochinchinenin/ALDH1A1 复合物,两者主要通过氢键作用、疏水作用、pipi-共轭作用发生结合。例如小分子Cochinchinenin 和ALDH1A1 蛋白上的TRP-178、GLY-125、ASN-121、GLU-269、TYR-297 形成氢键作用,和ALA-462、TRP-128、LYS128、VAL-174、PHE-171、TYR-297、ILE-304、VAL-460、PHE-466 形成疏水作用。此外,小分子Cochinchinenin 还和蛋白上的PHE-171 形成pipi-共轭作用。见图4C。对于Herpetone/ALDH1A1 复合物,小分子Herpetone 和蛋白上的TRP-178、THR-129、ASN-121 形成氢键作用,和VAL-174、TYR-297、PHE-290 发生疏水作用,和TYR-297、HIS-293 形成pipi-共轭作用。见图4D。进一步,对上述四组复合物进行分子动力学模拟,来分析这些复合物的结合稳定性以及更为精确的结合能。

图4 基于对接获得的小分子2,3-Dihydroheveaflavone、Sotetsuflavone、Cochinchinenin、Herpetone 与ALDH1A1 蛋白的结合模式,左图为整体视图,右图为局部视图,图中蓝色stick 为小分子,淡青色Cartoon 为蛋白,氢键作用以黄色虚线显示,疏水作用以灰色虚线显示,pipi-共轭作用以绿色虚线显示
2.3 稳定性分析 分子动力学模拟的均方根偏(RMSD)可以反映复合物的运动过程,RMSD 越大以及波动越剧烈表示运动剧烈,反之运动平稳。见图5。ALDH1A1 蛋白在未结合药物的情况下(黑线)模拟过程中波动程度大,而结合了药物后波动程度呈现更稳定的趋势,尤其是在ALDH1A1/TNP-004802、ALDH1A1/TNP-000924 复合物,它们在分子动力学模拟过程中RMSD 基本上在3 Å 以内稳定波动。暗示ALDH1A1/TNP-004802、ALDH1A1/TNP-000924 复合物结合非常稳定。

图5 分子动力学模拟过程中复合物RMSD 差随时间的变化
均方根波动(RMSF)可以反映分子动力学模拟的过程中蛋白的柔性。通常药物结合蛋白后,蛋白柔性下降,进而达到稳定蛋白的作用,发挥酶活效果。除了蛋白两端以及130~145 段氨基酸外,其余各个序列段的RMSF 数值偏低,表示整个结构的柔性较低。对比结合化合物和未结合化合物蛋白的RMSF 数值,发现结合化合物时,蛋白的RMSF 更低,其中结合TNP-004802最显著,其次为TBB-02884、TNP-000924、TNP-001406。见图6。

图6 基于分子动力学模拟轨迹计算的RMSF
2.4 MM/GBSA 结果 在本研究中基于MM/GBSA 计算方法,对分子动力学模拟轨迹充分采样以准确计算小分子与蛋白的结合能。ALDH1A1 蛋白和TBB-02884、TNP-001406、TNP-004802、TNP-000924 的结合能分别为(-36.35±3.70)、(-43.61±2.32)、(-56.66±3.83)、(-52.26±2.64) kcal/mol。四个组合的结合能都非常高,其中ALDH1A1 蛋白和TNP-004802、TNP-000924 的结合能甚至>-50.0 kcal/mol,暗示其结合较好。通过残基分解,四个组合结合的主要贡献力为疏水作用,其次是静电作用,最后是非极性溶剂化自由能。见表3。
表3 MM/GBSA 计算结合自由能(,kcal/mol)

表3 MM/GBSA 计算结合自由能(,kcal/mol)
ΔEvdW: 范德瓦耳斯能量(van der Waals energy);ΔEelec: 静电能量(electrostatic energy);ΔGGB: 静电对溶剂化的贡献(electrostatic contribution to solvation);ΔGSA: 溶剂化的非极性贡献(non-polar contribution to solvation);ΔGbind: 结合自由能(binding free energy)
ALDH1A1 上的氨基酸类型对该结合起重要作用,对理解结合机制也非常重要,将4 个候选分子与ALDH1A1 结合的能量分解至每个残基的贡献。复合物中对结合能起贡献的Top-10 氨基酸:对于ALDH1A1/TBB-02884 复合物,ILE 304、PHE 289、PHE 170、ASN 121、TRP 178、TYR 297、VAL 460、TYR 457、CYS 302、VAL 459 为关键性氨基酸;对于ALDH1A1/TNP-001406 复合物,TRP 178、TYR 297、VAL 460、ASN 121、PHE 171、ILE 304、SER 461、GLY 125、LYS 128、ALA 462 为结合贡献大的氨基酸;对于ALDH1A1/TNP-004802 复合物,TRP 178、ILE 304、VAL 174、TYR 457、VAL 460、PHE 171、TYR 297、CYS 302、ASN 121、SER 461 为结合贡献大的氨基酸;对于TNP-000924 复合物,TYR 297、GLY 125、ALA 462、VAL 460、TRP 178、PHE 290、VAL 459、SER 461、GLN 463、VAL 174 为结合贡献大的氨基酸。见图7。
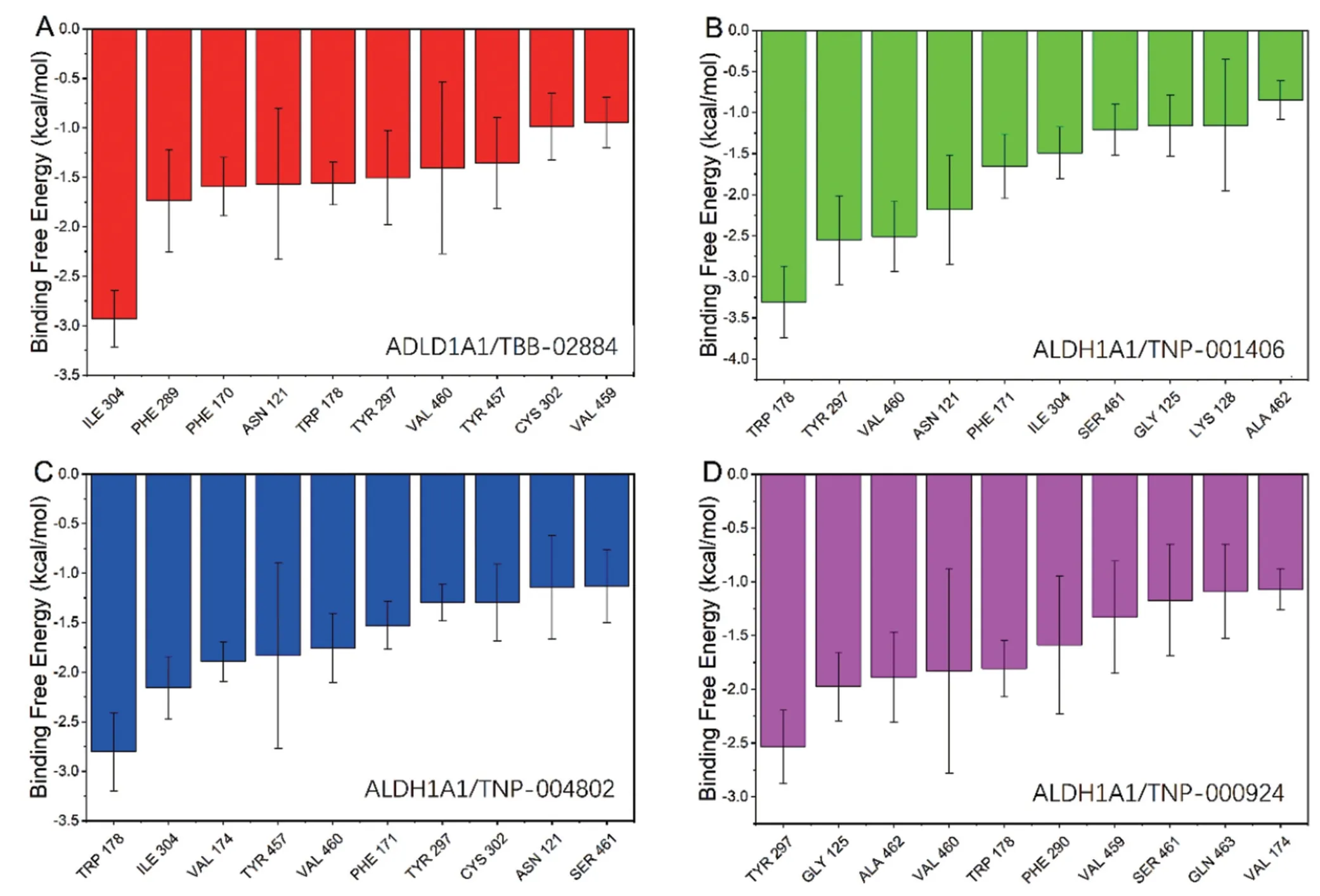
图7 对小分子和蛋白结合能起贡献的Top-10 氨基酸
2.5 氢键分析 氢键作用是小分子化合物和蛋白结合过程中最强的非共价作用之一,在分子动力学模拟期间,统计各个复合物形成的氢键情况:小分子TBB-02884、TNP-001406、TNP-004802、TNP-000924 和ALDH1A1 蛋白在模拟过程中形成的氢键数目都维持在2 个以上。表明氢键作用对四个复合物稳定存在起重要作用。TNP-001406、TNP-000924 在模拟过程中和ALDH1A1 蛋白形成的氢键频率要高于TBB-02884、TNP-004802,意味着TNP-001406、TNP-000924 和ALDH1A1 蛋白的结合更加依赖于氢键作用。见图8。
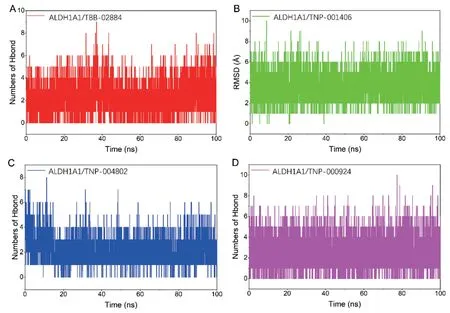
图8 分子动力模拟过程中小分子与蛋白的氢键数目变化
2.6 酶活抑制实验结果 TNP-004802 与ALDH1A1结合稳定,且结合能最高,进一步运用酶动力学实验方法比较TNP-004802 与NCT-501 对ALDH1A1 的抑制活性,结果显示,两者均有较好的抑制活性,低浓度的TNP-004802 抑制活性高于NCT-501。见图9。

图9 TNP-004802 与NCT-501 对ALDH1A1 的抑制活性
3 讨论
醛脱氢酶(ALDHs)是一类依赖于NAD(P)的酶,可催化醛氧化成羧酸。ALDHs 是四聚体酶,N 端的结合域与辅因子 NAD+结合,从而改变四聚体构象[15]。催化位点的亚基结构形成结合口袋与乙醛等底物结合后发生结构改变,这种相对柔性的结构改变是限制醛氧化的关键步骤[16]。人体中,已知有19 种功能性基因编码ALDHs 的基因[17]。视黄醛的主要催化酶是ALDH1A1。目前分离到的多种CSCs 发现ALDH1A1表达水平升高[18],联合CD133、CXCR4、Notch-4 等标志物用于分离和鉴定CSCs。因此,ALDH1A1 可作为抗肿瘤治疗的重要靶标,研发 ALDH1A1 抑制剂是目前肿瘤治疗领域的热点,目前已发现多种不同结构的抑制剂,未来研究方向将主要在先导化合物的优化。许多肿瘤细胞系以及患者中ALDH1A1 酶水平升高导致对化疗和放疗的耐药[19]。
ALDHs 抑制剂双硫仑适合从肿瘤中根除化学抗性干细胞样肿瘤起始细胞和预防复发的治疗方法[20]。Omran[21]开发了双硫仑衍生物2b 显示出与双硫仑对ALDH1A1 相当的活性。Verma 等[22]通过三维定量构效关系和骨架跃迁设计了21 种ALDH1A1 抑制剂,全部化合物均显示半抑制浓度(IC50)值在0.02~0.80 μM的范围内,表明这些作为环磷酰胺耐药的辅助治疗的潜力。研究表明[23],乙醛脱氢酶1 的催化活性中心主要为半胱氨酸-302(Cys302),已报道的抑制剂主要通过与Cys302 结合导致酶的催化活性受抑制。本研究对17580 个中药分子进行Lipinski Ro5 规则、分子对接、ADME 预测,选取了4 个化合物进行分子动力学模拟,Cochinchinenin、Herpetone、2,3-Dihydroheveaflavone、Sotetsuflavone。本结果发现Cochinchinenin 与ALDH1A1的Cys302、TRP 178 等多个氨基酸残基位点结合,稳定性最显著,结合能最高,酶活抑制实验显示了其对ALDH1A1 良好的抑制活性。
Cochinchinenin 是来源于中药血竭的化合物,《唐本草》论及血竭的功效为“主五脏邪气,带下,止痛,破积血,金创生肉”,常用于跌打损伤、恶疮、瘰疬等。血竭一直以来用于恶性肿瘤的治疗,明代龚廷贤所著《万病回春·积聚》提及含血竭的药方”神化丹”之功效:“消癖积、破血块、下鬼胎、通经脉及诸痞积血气块”。现代临床应用单味血竭粉外敷、内服治疗癌性溃疡108 例,取得满意疗效[24],外敷可用于癌性疼痛[25],实验研究方面,血竭素高氯酸盐通过降低细胞线粒体膜电位,诱导人乳腺癌细胞MCF-7 凋亡[26];通过活化MAPK 通路中的JNK 和p38 发挥促进胶质瘤细胞LU251 凋亡的作用[27]。
本研究虚拟筛选血竭素作为ALDH1A1 抑制剂,具有抑制肿瘤干细胞增殖和耐药的可能药效,血竭素可作为先导化合物研究,进一步可开展药物化学、药理学及临床研究,找到与ALDH1A1 相关的抑制癌症干细胞增殖和耐药的有效药物。
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
大学物理(2022年9期)2022-09-28 01:10:52
昆明医科大学学报(2021年1期)2021-02-07 01:06:14
物理通报(2020年7期)2020-07-01 09:28:02
中成药(2018年5期)2018-06-06 03:12:14
原子与分子物理学报(2015年3期)2015-11-24 12:49:34
文理导航(2015年26期)2015-09-29 14:12:24
中国生化药物杂志(2015年4期)2015-07-07 12:06:06
中国中医药现代远程教育(2014年16期)2014-03-01 04:28:34
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
