
利用WGCNA鉴定玉米非生物胁迫相关基因共表达网络
2023-01-16 03:43:42蒋环琪程丽沙李曼菲杜何为
作物学报 2023年3期
邓 照 蒋环琪 程丽沙 刘 睿 黄 敏 李曼菲 杜何为
利用WGCNA鉴定玉米非生物胁迫相关基因共表达网络
邓 照**蒋环琪**程丽沙 刘 睿 黄 敏 李曼菲*杜何为*
长江大学生命科学学院, 湖北荆州 434025
加权基因共表达网络分析(weighted gene co-expression network analysis, WGCNA)是一种经典的系统生物学分析方法, 可用来鉴定协同表达的基因模块, 探索模块与目标性状的生物学相关性, 并挖掘模块网络中的核心基因。本文收集了低温胁迫、高温胁迫、干旱胁迫和盐胁迫处理下玉米(L.)根、茎、叶等组织的58份转录组数据, 利用WGCNA方法鉴定玉米非生物胁迫的基因共表达网络模块。过滤转录组数据中12,552个低表达基因后, 利用余下27,204个高表达基因构建共表达网络, 分析得到25个模块。根据玉米中已报道非生物胁迫相关基因与差异表达基因在模块中的分布, 筛选出与低温胁迫、高温胁迫、干旱胁迫和盐胁迫最相关的mediumpurple4、ivory、coral2、darkseagreen4模块和响应多种胁迫的green模块。随后对这5个模块的基因进行GO富集分析, 发现在这些模块内与非生物胁迫相关基因的在非生物胁迫调控相关功能显著富集, 如胁迫响应、过氧化物酶活性等。对这5个模块作相关性分析, 预测出、、、和等10个非生物胁迫相关的核心基因。本研究为玉米非生物胁迫相关基因的挖掘和非生物胁迫调控网络研究等提供了新思路。
玉米; 非生物胁迫; WGCNA; 核心基因
随着高通量测序技术的飞速发展, 海量的测序数据应运而生。加权共表达网络分析(weighted gene co-expression network analysis, WGCNA)利用植物体内生命活动互相关联的特点, 结合高通量测序技术获得基因的表达量数据将具有相似表达模式的基因聚类到同一个模块。WGCNA对基因间的连接系统进行幂次处理, 使模块内部的基因符合无尺度网络拓扑结构。网络中少数基因与大多数基因存在联系, 通过相关性分析, 可以预测位于中心的少数基因, 这些少数基因可能为网络的关键调控基因[1]。WGCNA多被用于研究共表达网络与植物性状之间的生物学关系, 挖掘与性状之间高度关联的关键基因。自2008年Langfelder和Horvath[2]提出加权共表达网络分析以来, 通过该方法寻找到了许多与植物表型性状、响应生物或非生物胁迫等方面的关键基因。如Kuang等[3]以香蕉果实为研究对象, 基于WGCNA构建了乙烯调控香蕉果实成熟的转录因子调控网络, 获得了25个关键转录因子, 这些转录因子能通过调控下游成熟相关基因的表达参与果实成熟过程; Sun等[4]利用绿色纤维棉花和白色纤维棉花之间的差异表达基因, 通过WGCNA揭示绿色色素积累相关的模块, 鉴定出了56个核心基因, 其中包括的2个同源基因,参与绿色色素的生物合成; Greenham等[5]利用WGCNA的方法, 分析了芸薹属作物在干旱胁迫响应初期的转录组变化, 鉴定出6个干旱胁迫抗性的核心基因, 包括细胞响应调节剂3 ()质体特异性核糖体蛋白6 ()生长素极性输出载体3 ()等; 马娟等[6]利用不同品种玉米在2种种植密度下的转录组数据, 结合WGCNA方法构建两种密度条件下的共表达网络, 鉴定到与株高和穗位高显著且高度相关的共表达模块15个, 其中两性状相同的模块共6个, 以已报道株高基因为核心, 构建基因网络, 发现生长素转录因子、、、光合系统II氧进化多肽和光合系统IN亚基等与核心基因存在关联。
玉米作为我国第一大粮食饲料作物, 在农业生产和国民经济发展中占有重要地位。玉米从北纬58° (加拿大和俄罗斯)至南纬40° (南美), 都有广泛的种植。受到全球气候变暖的影响, 各种反常气候出现频率增加, 玉米遭受低温、高温、干旱和盐等非生物胁迫的可能性增高。植物遭受非生物胁迫后生长发育缓慢, 严重时会导致死亡[7]。非生物胁迫会使植物产生应激反应, 应激反应会破坏植物细胞结构和代谢稳态, 降低农作物的产量和品质[8]。植物对非生物胁迫的耐受性受多基因控制[9], 生物信息技术的发展和基因工程改造手段的进步为研究植物响应非生物胁迫相关基因提供了可能[10]。
本研究利用玉米不同组织部位在低温胁迫、高温胁迫、干旱胁迫和盐胁迫处理下获得的共58份转录组数据[11-18], 构建WGCNA共表达网络, 分析得到25个模块。从玉米遗传和基因组数据库(https:// maizegdb.org/)获取已报道的与低温、高温、干旱和盐相关的44个基因, 通过查看已报道基因在模块中的分布, 结合玉米不同胁迫处理下差异表达基因在模块中的占比, 筛选出与各胁迫最相关的模块进行功能富集分析。通过对胁迫相关模块的相关性系数分析, 预测模块中核心基因并进行功能注释。本研究为玉米非生物胁迫基因的网络研究和非生物胁迫的分子育种提供了理论基础。
1 材料与方法
1.1 玉米转录组数据的获取与分析
玉米低温胁迫、高温胁迫、干旱胁迫和盐胁迫处理的58个转录组数据来源于NCBI的SRA (Sequence Read Archive)数据库。首先利用SRAtoolkit[19]软件(version 2.9.6)的fastdump将SRA格式文件转换为序列文件fastq, 利用FastQC[20]软件(version 0.11.9)[21]对原始测序数据进行质量评估, 查看网上下载数据接头序列是否去除、是否有其他物种DNA污染、是否去除重复序列等, 紧接着用Fastp软件(version 0.23.0)去除接头、过滤低质量的reads, 得到clean data, 参数为默认参数, 滑动窗大小–W 4, 平均质量–M 20, 短序列过滤–l 15, 质量过滤–q 15。利用Hisat2[22]软件(version 2.2.1)将clean data比对到玉米基因组(B73_RefGen_v5)。玉米基因组序列和注释信息下载自MaizeGDB (https://maizegdb.org/)数据库。用featureCounts[22]获得数据的reads计数后, 利用R软件(R version 4.1.2)计算基因表达的TPM值(transcripts per million), 选取58个样品中至少有一组TPM值大于10的基因构建表达矩阵, 用于基因共表达网络的分析。
1.2 加权基因共表达网络的构建
基因的表达量矩阵来自于不同非生物胁迫处理下的基因表达量, 使用R软件WGCNA包(version 1.6.6), 构建加权基因共表达网络[2]。为了确保网络符合无尺度网络分布, WGCNA需要使用合适的加权系数β。使用WGCNA包中的pickSoftThreashold计算权重值, 先设置β=1–30, 分别计算其对应的相关系数和基因连接度均值, β的选取标准为满足相关系数的平方接近0.8, 同时还保证一定的基因连接度。根据图1的结果所示, β选取15来构建共表达网络, 使用blockwiseModules构建无尺度网络, 参数按照默认设置[2]。
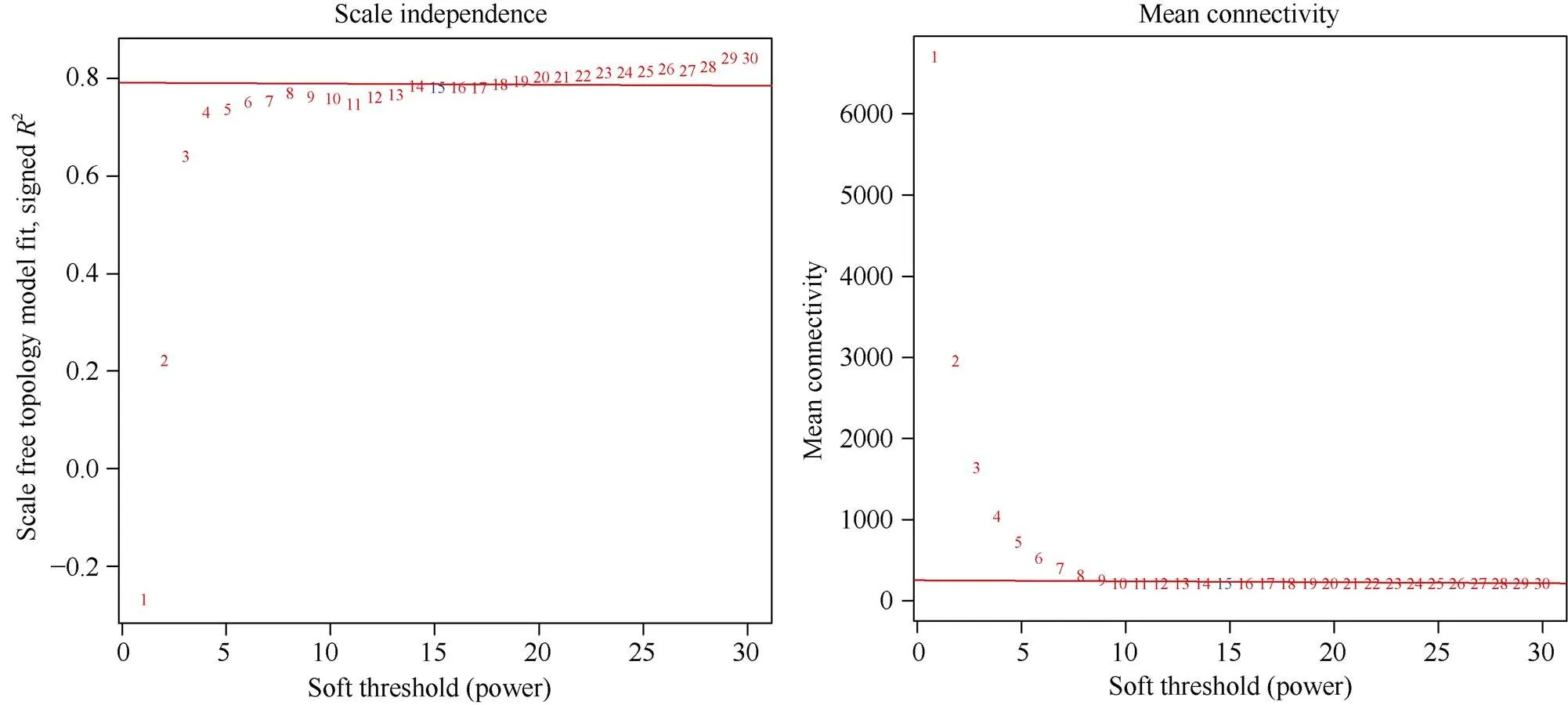
图1 软阈值的选择
1.3 特异性模块的筛选
选取玉米已报道的非生物胁迫基因, 确定已报道基因所在模块, 并利用Cufflink (version 2.2.1)[24]分别对数据库下载中下载到低温胁迫、高温胁迫、干旱胁迫和盐胁迫处理的玉米幼苗组织进行差异表达分析, 使用玉米参考基因组B73_RefGen_v5, 分析差异基因所在的表达模块, 确定响应非生物胁迫反应的特异性模块。
1.4 模块基因的富集分析
首先提取出组织特异性模块中的基因, 再将模块基因提交到agriGO (http://systemsbiology.cau.edu. cn/agriGOv2/specises_analysis.php?SpeciseID=20&latin=Zea_mays)在线分析工具, 采用单一富集分析(singular enrichment analysis, SEA)法进行富集分析, 利用Fisher's校验和Bonferronni多重校验来进行显著GO类型的筛选[25]。利用Cytoscape (version 3.9.1)对模块中的网络进行可视化[26], 筛选出核心基因。核心基因选取主要通过该基因与其余相关性或其在调控网络中的位置确定的, 如果该基因与更多基因的相关性超过阈值, 或者该基因在调控网络中的节点位置, 则该基因为核心基因, 相关系数的阈值为>0.95。
1.5 核心基因的表达分析
将B73的种子种植于10个塑料杯中, 每杯10粒种子, 待幼苗长至二叶一心期, 分别取2杯进行低温、高温、干旱和盐胁迫处理各6 h, 剩余2杯作为对照。其中低温胁迫和高温胁迫分别对实验组进行4℃和38℃处理6 h, 对照组室温(冬季组培室温度约22℃)生长, 胁迫处理后取叶片提取RNA; 干旱胁迫和盐胁迫分别对实验组用10% PEG溶液和150mmol的NaCl溶液各取100 mL于烧杯中浸没幼苗根部6 h, 对照组用无菌水100 mL浸没根部6 h, 胁迫后取根部提取RNA。将提取的RNA反转录为cDNA, 以核心基因的cDNA参考序列为模板, 设计qPCR引物, 以为内参基因, 利用荧光定量PCR实验结果计算核心基因的相对表达量, 分析核心基因在实验组和对照组中的相对表达量差异, 验证核心基因是否响应对应非生物胁迫。相对表达量的计算方法为:

2 结果与分析
2.1 玉米加权基因共表达网络构建
根据TPM值, 将玉米表达矩阵中表达量较低的12,552个基因进行过滤, 获得27,204个高表达基因。选择加权值β=15来构建无尺度网络, 之后按照混合动态剪切的标注划分模块, 随之依次计算每个模块的特征向量值(eigengenes), 对距离较近的模块进行合并, 最终获得25个模块, 每个模块用不同的颜色来表示(图2)。其中magenta模块聚类到的基因数目最多, 为10,172个, mistyrose模块包含的基因最少, 为44个。
文献[27-56]中已报道非生物胁迫相关基因共44个(附表1), 分布在12个不同模块中(图3)。其中已报道的低温胁迫相关基因共有3个, 分别位于brown4、darkviolet和green模块中; 已报道的高温胁迫相关基因在共表达网络中共有15个, ivory模块中存在最多, 为7个, 其余8个分别存在于coral2、magenta和darkseagreen3模块等5个模块; 已报道的干旱胁迫相关基因在共表达网络中有14个, 其中magenta、coral2和darkseagreen4模块各存在3个, 其余5个分布于brown4、coral1、darkviolet和green模块; 已报道的盐胁迫相关基因共有19个, 在magenta模块中存在最多, 为7个, 其余12个分布于brown4、coral1、coral2等10个模块。
为了验证模块选取的准确性, 对数据中低温胁迫、高温胁迫、干旱胁迫和盐胁迫处理的玉米幼苗组织的24份转录组数据进行了差异表达基因分析, 其中低温胁迫和高温胁迫选择的是自交系B73在胁迫处理下的转录组数据, 而干旱胁迫和盐胁迫则分别选取干旱胁迫敏感型自交系和盐胁迫敏感性自交系在胁迫处理下的转录数据。mediumpurple4模块中低温胁迫差异表达基因占比为72%, 而含有已报道低温胁迫相关基因的brown4、darkviolet和green模块中低温胁迫差异表达基因数目只占模块基因总数目的35%、40%、48%, 因此mediumpurple4模块与低温胁迫最相关。ivory模块中高温胁迫差异表达基因数目占75%, 其余5个含有已报道高温胁迫相关基因的模块中darkseagreen3模块占比为48%, 其余模块分别占比28%、25%、18%和10%, 因此ivory模块与高温胁迫的相关性最高。green模块中干旱胁迫差异表达基因占比为22%, 而coral2、darkseagreen4和magenta模块中干旱胁迫差异表达基因数目分别占该模块基因总数的19%、6%和3%, 因此coral2模块与干旱胁迫最相关。darkseagreen模块中盐胁迫差异表达基因数目占该模块基因总数的61%, 其余10个含有已报道盐胁迫相关基因的模块中magenta模块占比最高为11%, 因此darkseagreen4模块与盐胁迫最相关(图4)。
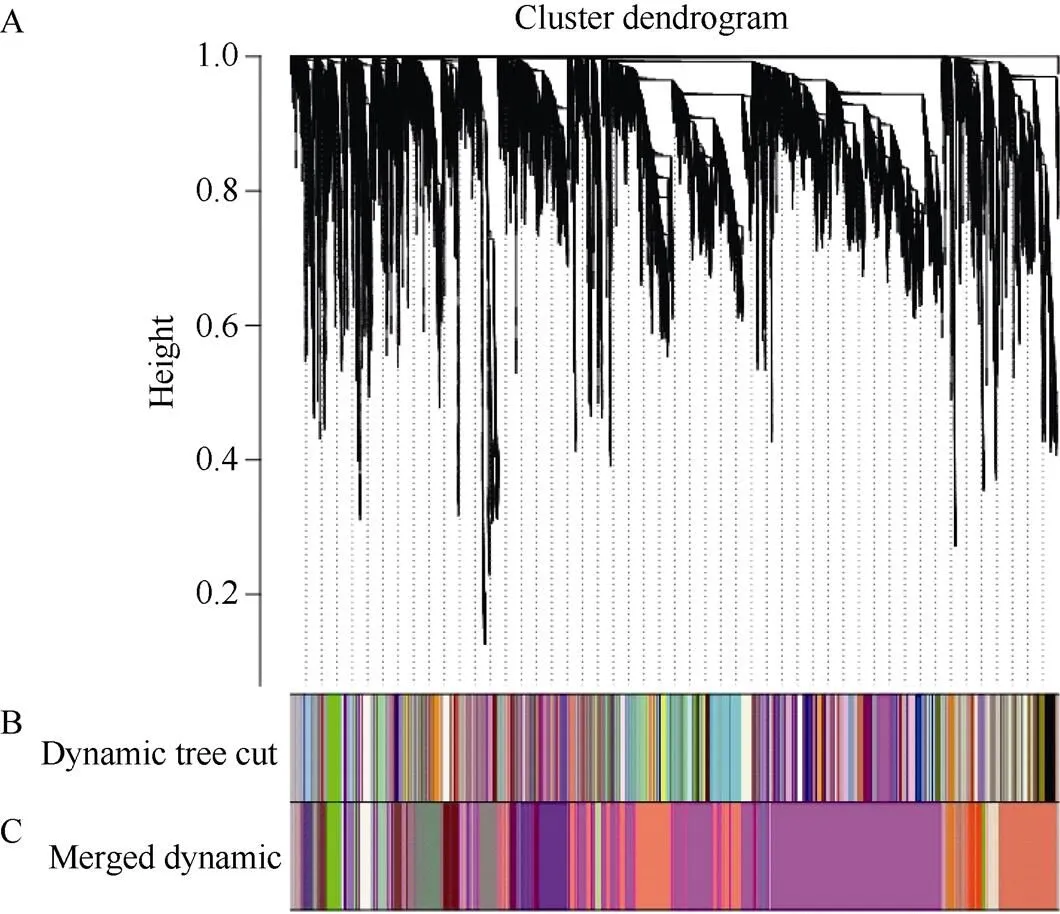
图2 基因聚类树和模块分割
A: 样品的参差聚类树; B: dynamic tree cut表示根据各个基因的表达量计算划分的模块; C: merged dynamic是根据dynamic tree cut合并相似模块的结果。
A: the hierarchical clustering tree of samples; B: the dynamic tree cut represents the module divided according to the expression of each gene; C: the merged dynamic is the merging similar modules according to the dynamic tree cut.
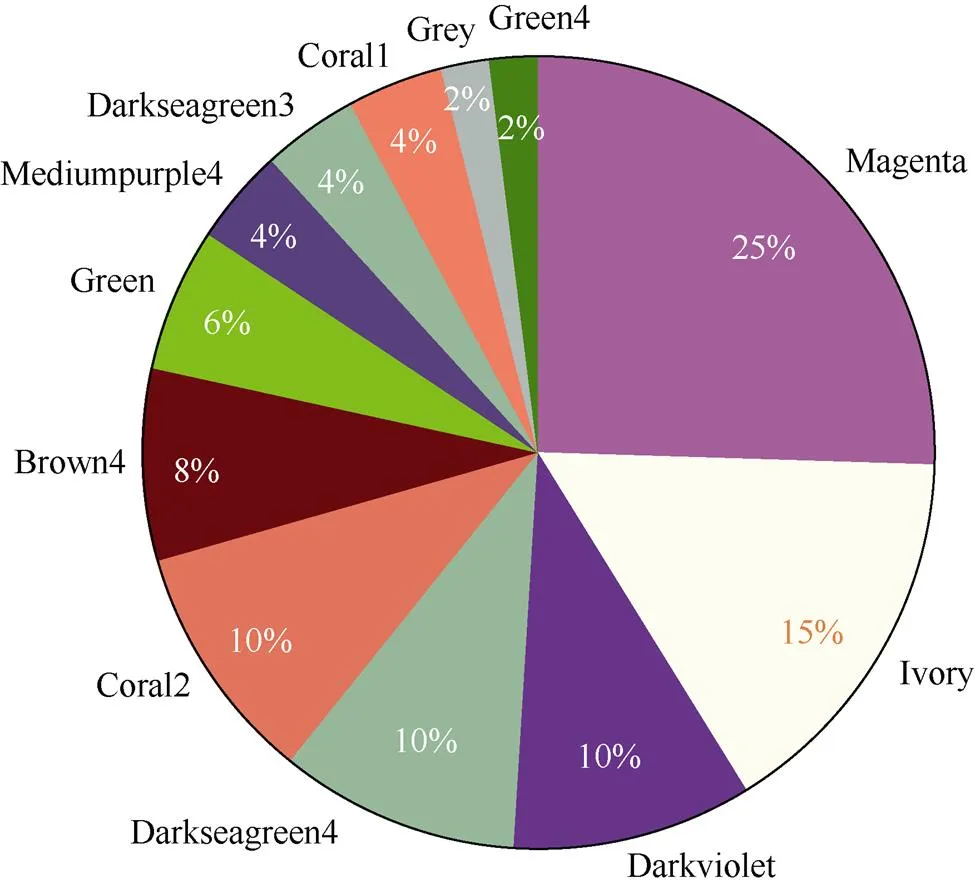
图3 报道基因分布情况
图中英文表示模块颜色, 百分数表示各模块已报道非生物胁迫基因数与已报道基因总数的比值。
The English words in the figure indicate the module colors, and the percentage indicates the ratio of the number of reported abiotic stress genes to the total number of reported genes in each module.
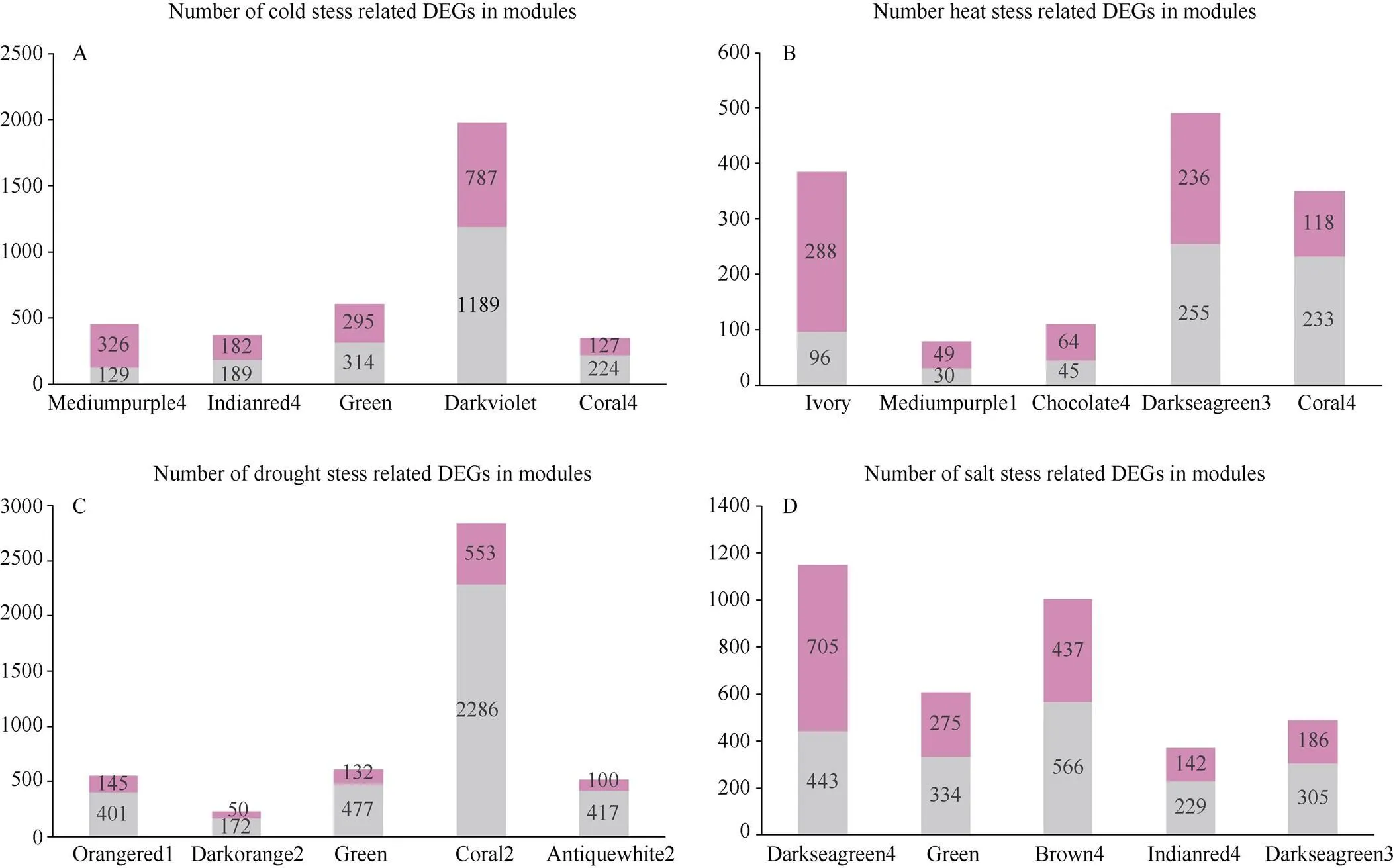
图4 各模块中胁迫相关差异表达基因的数目
横坐标为模块颜色, 纵坐标为基因个数, 粉色部分表示差异表达基因个数, 灰色部分表示模块中非差异表达基因的个数。A: 低温胁迫差异表达基因占比最多的5个模块; B: 高温胁迫差异表达基因占比最多的5个模块; C: 干旱胁迫差异表达基因占比最多的5个模块; D: 盐胁迫差异表达基因占比最多的5个模块。
The abscissa in the figure is the module colors, the ordinate is the number of genes, the pink part represents the number of DEGs, and the gray part represents the number of non-differentially expressed genes in the module. A: five modules with the highest proportion of DEGs in cold stress. B: five modules with the highest percentage of DEGs in heat stress. C: five modules with the highest percentage of DEGs in drought stress. D: five modules with the highest percentage of DEGs in salt stress.
2.2 模块的GO富集分析
选取存在已报道基因的模块, 结合不同胁迫下差异表达基因在该模块中的分布情况, 确定mediumpurple4模块代表低温胁迫相关模块, ivory模块代表高温胁迫相关模块, coral2模块代表干旱胁迫相关模块, darkseagreen4模块代表盐胁迫相关模块。并对这些模块进行GO功能富集分析, 富集结果涉及到了生物学过程(biological process, BP)、分子功能(molecular function, MF)和细胞组分(cellular component, CC)。
低温胁迫相关的mediumpurple4模块富集到了与低温相关的调控通路, 如昼夜节律(GO:0007623)、脱落酸激活的信号通路(GO:0009738)、温度刺激的响应(GO:0009266)等。高温胁迫相关的ivory模块富集到了高温响应(GO:0009408)、胁迫响应(GO: 0006950)、刺激响应(GO:0050896)等功能。干旱相关的coral2模块富集到了氧化还原过程(GO:0055114)、分解代谢过程(GO:0009056)、氧化应激响应(GO:0006979)等功能。盐胁迫相关的darkseagreen4模块富集到了有机酸分解代谢过程(GO:0016054)、氧化还原过程(GO:0055114)、对外源刺激的响应(GO:0009605)等胁迫响应的功能(表1和附表2)。
分别对低温胁迫、高温胁迫、干旱胁迫和盐胁迫相关的4个模块进行相关性分析, 根据模块内基因的表达量计算与模块内所有基因的pearson相关系数, 根据pearson相关系数, 使用Cytoscape软件对网络进行可视化(图5和表2), 找到表达网络中的核心基因。发现mediumpurple4模块中的和; ivory模块中的和; coral2模块中的和; darkseagreen4模块中的和处于网络的核心位置(图5)。其中在mediumpurple4的核心基因为一种MYB相关转录因子(),编码单加氧酶; ivory模块中Zm00001eb423300为热休克蛋白(HSP20),编码HSP ATPase的激活剂; coral2模块中Zm00001eb265310是类细胞壁相关激酶受体蛋白激酶(wall associated kinase recetor-like kinase, WAK-RLK), Zm00001eb058250为一种过氧化物酶(POD16); darkseagreen4模块中Zm00001eb375120为2-酮戊二酸(2OG)和Fe (II)依赖性加氧酶超家族蛋白(GPM626), Zm00001eb323090为异柠檬酸裂解酶1 (ICL1)。
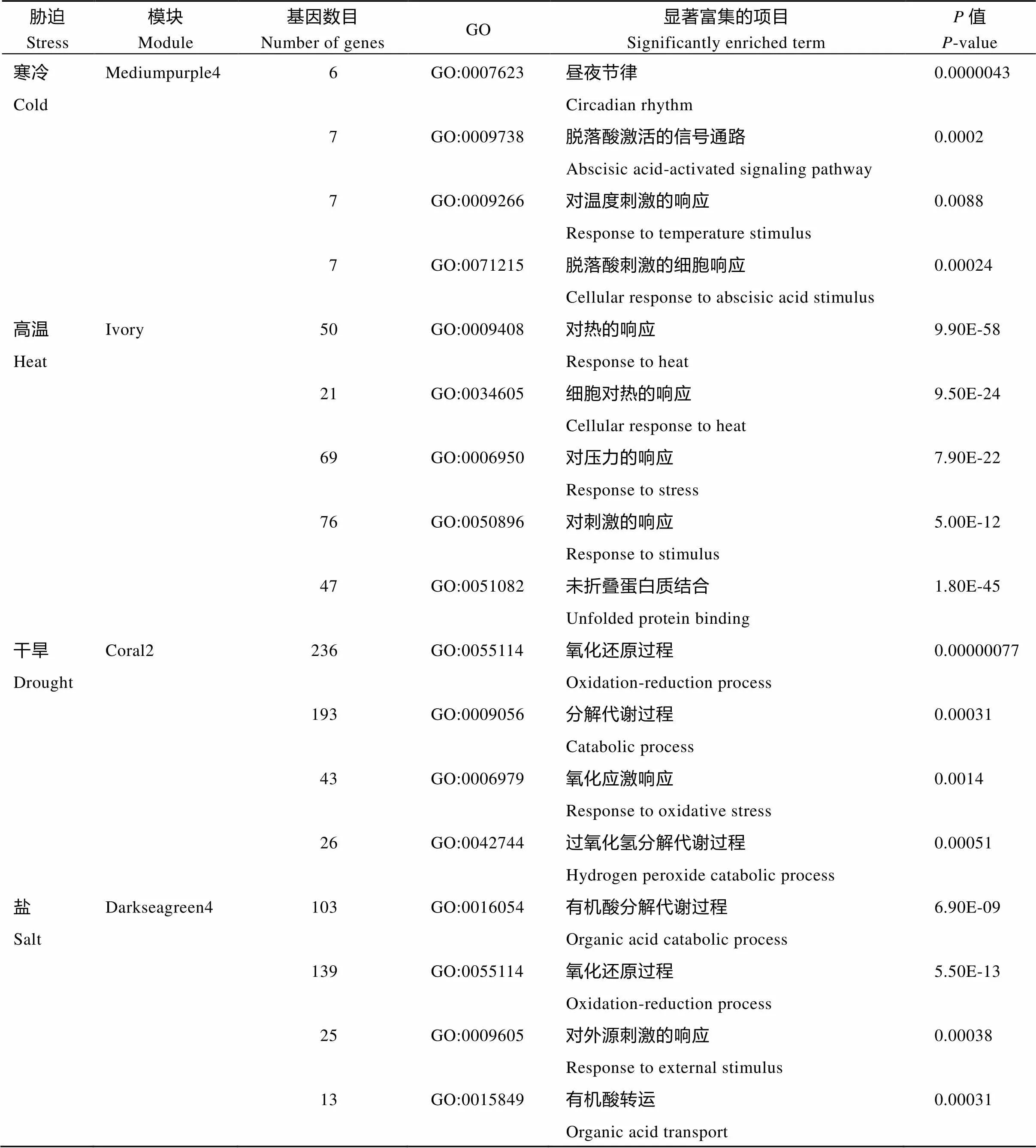
表1 模块GO富集情况(部分)
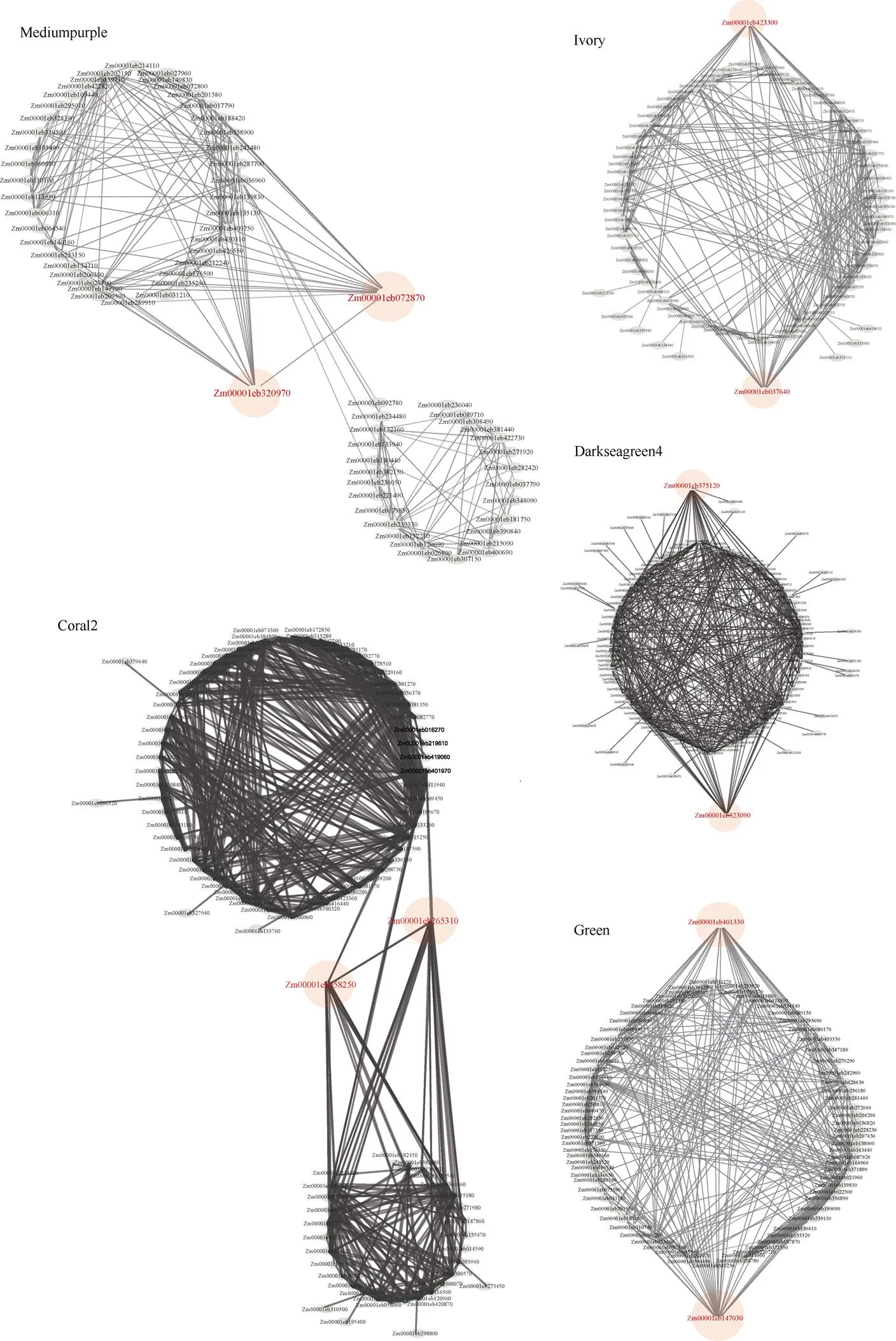
图5 非生物胁迫相关的共表达网络
标红色的基因代表该共表达网络的核心基因。
The genes marked in red represent the core genes in the co-expression network.

表2 模块核心基因功能注释
2.3 同时响应多种非生物胁迫模块的GO富集分析
同时响应低温胁迫、高温胁迫、干旱胁迫和盐胁迫这4种非生物胁迫的差异表达基因共有226个(图6), 其中darkseagreen4模块聚类了41个基因, 数目最多; green模块聚类了33个基因; magenta模块聚类了28个基因(图7)。darkseagreen4模块被定义为盐胁迫相关模块, 因而我们不考虑其为响应多种非生物胁迫的模块。结合模块中非生物胁迫差异基因的占比, magenta模块为0.3%, green模块为5.4%, 我们认为green模块为同时响应4种非生物胁迫相关的模块。对green模块做GO富集分析, 发现green模块富集到了钙离子结合(GO:0005509)、钙调蛋白结合(GO:00055)、有机酸跨膜转运(GO:1903825)等响应非生物胁迫的功能(表3和附表3)。对green模块做相关性系数分析, 并使用Cytoscape软件对网络进行可视化, 其中0和处于网络的中心位置(图5),编码类富含半胱氨酸和组氨酸丰富域(CHP)的锌指蛋白,0是NAC转录因子()。
2.4 转录因子在模块中的功能富集分析
转录因子(transcription factor, TF)也称为反式作用因子, 是指能够与真核基因的顺式作用元件发生特异性相互作用, 并对基因的转录有激活或抑制作用的DNA结合蛋白。TF在植物生长发育和非生物胁迫防御反应等过程中具有重要调控作用, 因此我们对4个胁迫最相关模块内聚类的TF结合差异表达基因进行了分析。其中低温胁迫相关的mediumpurple4模块中聚类了26个差异表达的转录因子, 高温胁迫相关的ivory模块中聚类了15个差异表达的转录因子, 干旱胁迫相关的coral2模块中聚类了27个差异表达的转录因子, 盐胁迫相关的darkseagreen4聚类了37个差异表达的转录因子(附表4)。其中mediumpurple4模块中存在一个已报的bZIP家族转录因子, 该基因受高温诱导表达, 对玉米响应高温胁迫十分重要, 但目前还不清楚该基因是否还响应其他非生物胁迫[37]; 在ivory模块中存在2个已报道的Heat shock factor (HSF)家族转录因子, 其中在拟南芥中过表达后, 显著上调了热特异性HSP基因和胁迫相关基因的表达,超表达株系的耐热性、耐盐性和脱落酸敏感性显著增强[42]; darkseagreen4模块中的MYB家族转录因子, 在4种非生物胁迫的应激处理下都显著上调表达。在拟南芥中超表达能促进了转基因植株对盐胁迫的耐受性; 同时超表达也能增加许多非生物胁迫相关基因的表达, 使植物增强应对逆境胁迫的能力[53]; 另外, 在darkseagreen4模块中有一类NAC家族转录因子家族基因, 在玉米中超表达, 可以显著提高苗期玉米的抗旱能力, 增强玉米幼苗对水分的利用率, 并诱导干旱胁迫响应基因的上调表达[32]。
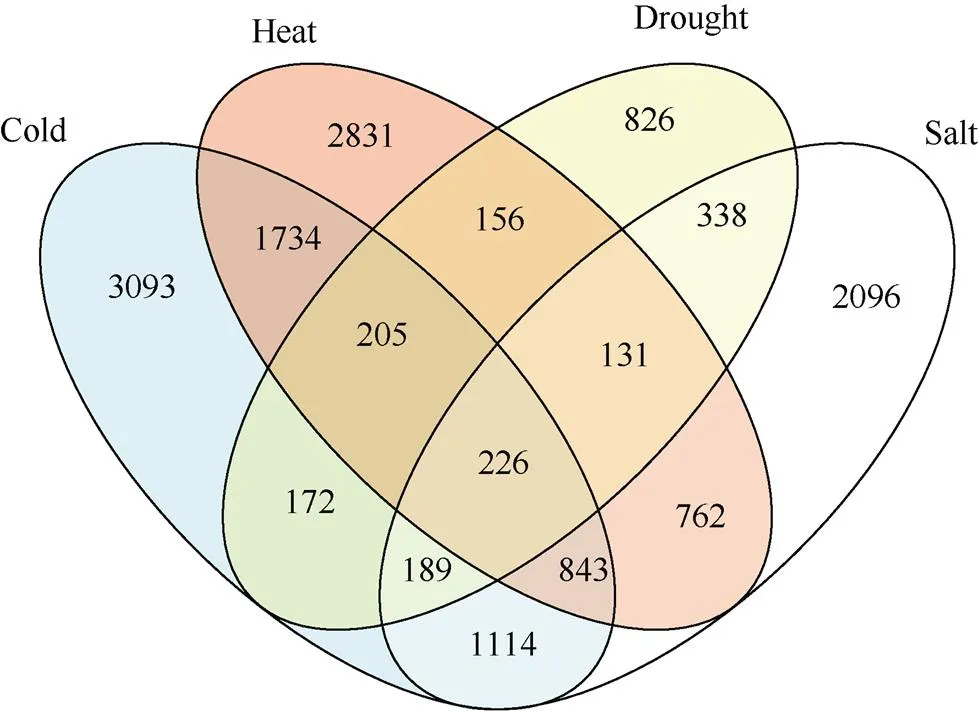
图6 非生物胁迫的差异表达基因
图中英文表示胁迫类型, 数字表示差异表达基因个数。
The words in the figure represent the stress types, and the numbers represent the number of differentially expressed genes.

图7 共表达基因在模块中的分布
图中数字代表共表达基因个数, 5个部分分别代表基因个数较多的4个模块和剩下的基因所占的比例。
The numbers in the figure represent the number of co-expressed genes, and the five parts represent the proportion of the four modules with a larger number of genes and the remaining genes.
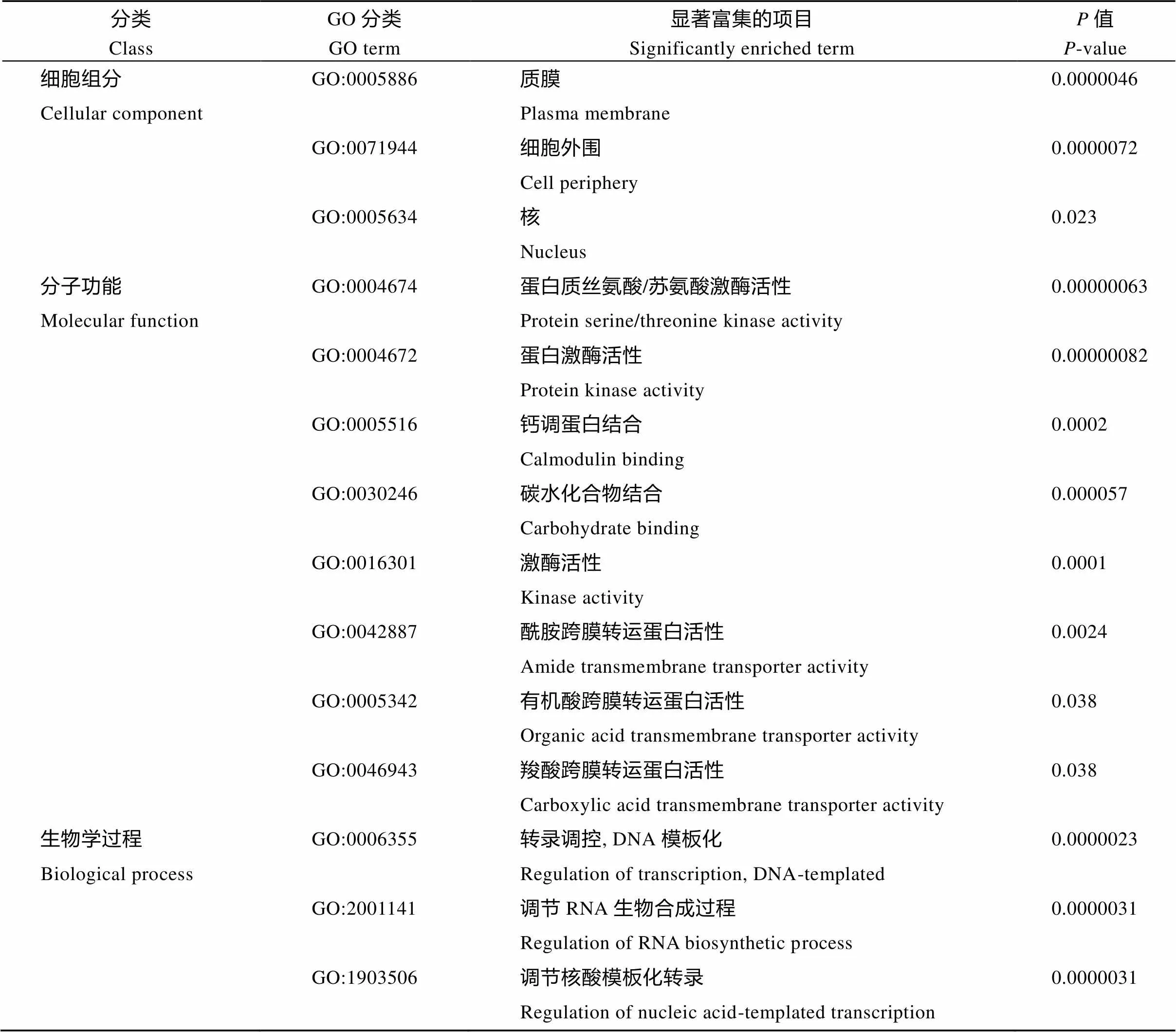
表3 Green模块GO富集情况(部分)
(续表3)
2.5 核心基因的表达验证
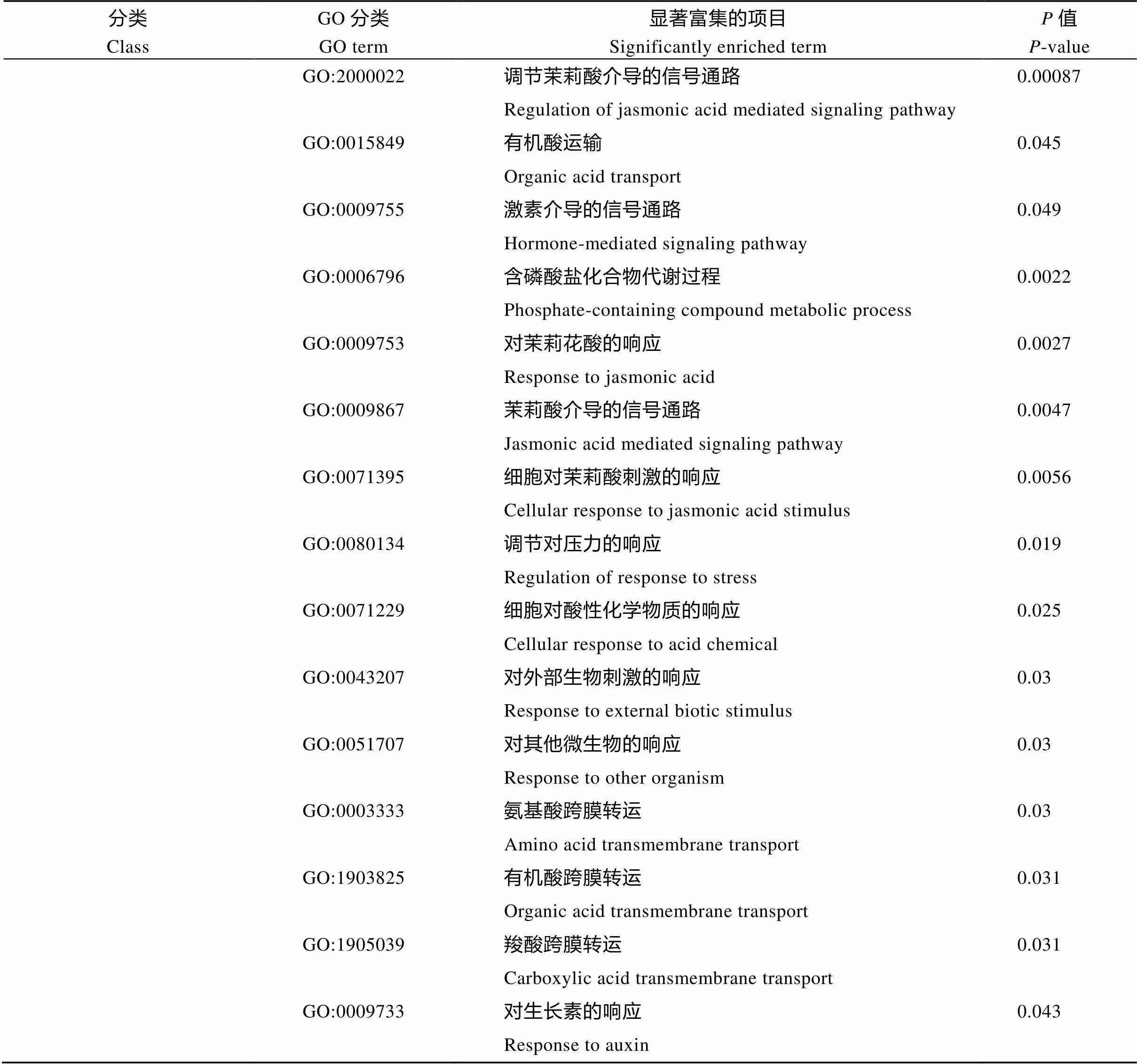
以核心基因cDNA参考序列为模板, 共设计出10对qPCR引物(附表5), 以B73材料不同非生物胁迫处理后提取的RNA反转录的cDNA作模板进行荧光定量,为内参基因, 得到以下结果: 其中mediumpurple4模块中的受低温胁迫后表达量显著上调, 而受低温胁迫后表达量减少为对照的1/3; ivory模块中的受高温胁迫后表达量显著上调, 而受高温胁迫后表达量减少为对照的1/7; coral2模块中的和受干旱胁迫后相对表达量显著下调, 其中表达量降低为对照的1/5左右,表达量降低为对照1/2左右; darkseagreen4模块中的和受盐胁迫后相对表达量显著降低约为对照的1/2 (图8)。同时green模块中的和在4种非生物胁迫下都有显著上调或下调的变化, 受低温和高温胁迫后两者的相对表达量都显著下调, 受干旱和盐胁迫后两者的相对表达量都显著上调, 其中受干旱和盐胁迫后表达量都增加了10倍左右(图9)。
3 讨论
植物响应非生物胁迫受多基因调控, 目前已报道基因响应非生物胁迫的生理机制及调控网络尚不明确。本研究收集了低温、高温、干旱和盐胁迫处理下的58份转录组数据建立共表达网络, 划分了25个模块, 并确定了分别响应4种非生物胁迫的模块mediumpurple4、ivory、coral2、darkseagreen4以及同时响应4种胁迫的模块darkseagreen4、green。对这些模块进行GO富集分析发现, 低温胁迫相关的mediumpurple4模块、高温胁迫相关的ivory模块分别富集到了温度刺激响应(GO:0009266)和高温响应(GO:0009408); 干旱相关的coral2模块和盐胁迫相关的darkseagreen4模块都富集到了氧化还原过程(GO:0055114, GO:0006979)。在干旱和盐胁迫中, 活性氧(reactive oxygen species, ROS)被诱导大量积累, 打破植物细胞氧化还原状态的平衡, 植物通过感知ROS信号激活细胞内的氧化还原代谢, 调节细胞氧化还原平衡来响应非生物胁迫[57]。同时响应多种胁迫的green模块富集到了有机酸分解代谢过程(GO:0016054)和有机酸跨膜转运(GO:1903825)。有机酸除了参加光合作用和呼吸作用, 还可以作为代谢活性溶质, 调节渗透压, 平衡过多的阳离子[58]。说明有机酸能节植物细胞的渗透压来响应非生物胁迫。另外green模还块富集到了钙离子结合(GO:0005509)和钙调蛋白结合(GO:00055)等与钙离子代谢相关的生物学过程。Ca2+是植物的第二信使, 参与非生物胁迫信号级联途径, 是响应非生物胁迫的重要信号途径。钙调素(Calmodulin, CaMs)和钙调素样蛋白(calmodulin-like proteins, CMLs)能感受细胞质中Ca2+的浓度变化, 积极参与信号感知和传递, 并与相关的效应蛋白相互作用, 形成一个级联信号调控网络[59]。我们发现这些模块富集到的GO功能和样品的处理方式基本吻合, 证明了WGCNA方法对所选取的大数据分析具有较高的可靠性。
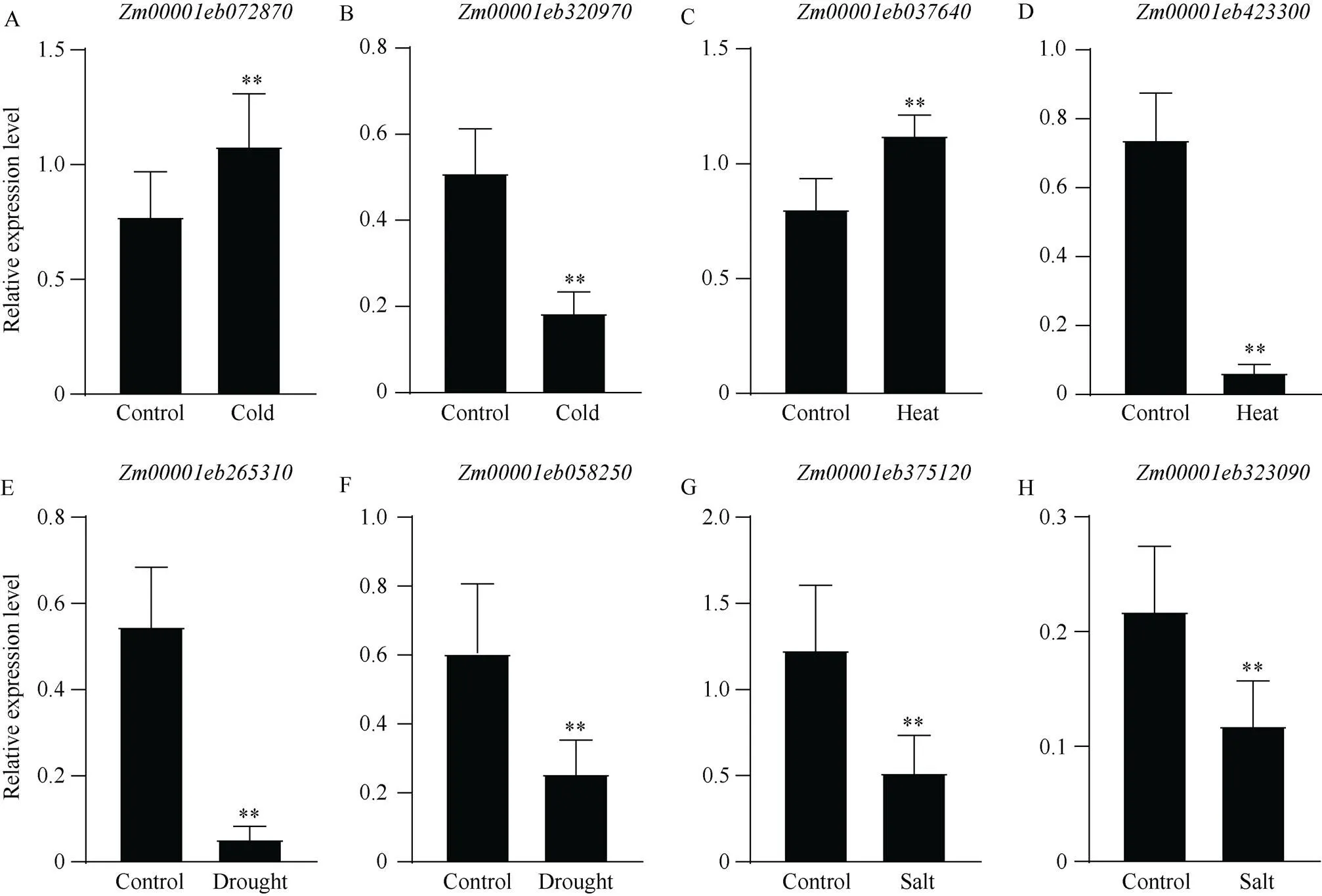
图8 非生物胁迫处理后核心基因的相对表达量
黑色柱子表示基因的相对表达量, X轴下方单词表示表示对应的处理。图A和图B表示低温胁迫后mediumpurple4模块中的和的相对表达量; 图C和图D表示高温胁迫后ivory模块中的Zm00001eb037640和Zm00001eb423300的相对表达量; 图E和图F表示干旱胁迫后coral2模块中的和0的相对表达量; 图G和图H表示盐胁迫后darkseagreen4模块中的0和的相对表达量。**:< 0.05。
The black bar in the figure represents the relative expression level of genes, and the words below the X-axis represent the corresponding treatments. A and B show the relative expression levels ofandin the mediumpurple4 module after cold stress; C and D show the relative expression levels ofandin ivory modules after heat stress; E and F represent the relative expression levels ofandin the coral2 module after drought stress; G and H show the relative expression levels ofandin darkseagreen4 modules after salt stress. **:< 0.05.
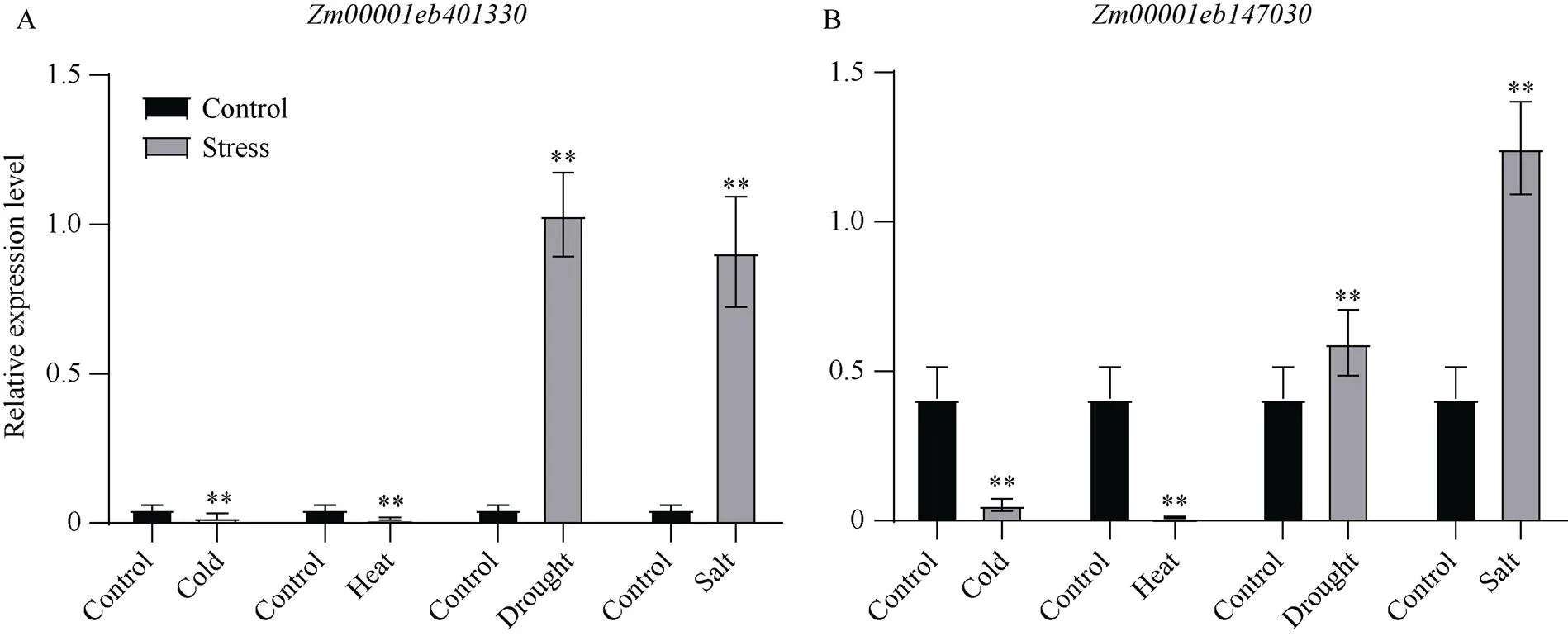
图9 同时响应多种非生物胁迫的核心基因相对表达量
图中黑色柱子表示正常处理下该基因的相对表达量, 灰色柱子表示非生物胁迫处理后该基因的相对表达量, X轴下方单词代表不同胁迫处理。图A和图B分别表示green模块中的和在低温胁迫、高温胁迫、干旱胁迫和盐胁迫下的相对表达量。**:< 0.05。
The black bar in the figure represents the relative expression levels of the genes under normal treatment, the gray bar represents the relative expression levels of the genes after abiotic stress treatment, and the words below the X axis represent different stress treatments. A and B represent the relative expression levels ofandin green module under cold stress, heat stress, drought stress, and salt stress, respectively. **:< 0.05.
上述模块中的10个核心基因, 8个是重要代谢过程中的调节酶或者含有特殊功能域的编码蛋白, 2个为转录因子。单加氧酶和过氧化物酶都是氧化还原过程中的关键酶, 而氧化还原过程是植物响应非生物胁迫的重要防御反应[60]; 热休克蛋白和HSP ATPase都是参与植物响应高温胁迫的重要调节蛋白[37,41-42]; 锌指蛋白在小麦中被报道可以增强盐胁迫的耐受性[61]; 类细胞壁相关激酶受体蛋白激酶(WAK-RLK)与植物细胞壁的果胶共价相联, 调节植物生长、参与植物信号转导和防御反应。WAK-RLK能介导细胞壁和细胞质的信号传导, 并通过级联信号途径调节下游效应蛋白, 来提高植物对生物和非生物胁迫的抗性[62]。2个转录因子分别是mediumpurple4模块中的和green模块中的, 这2个转录因子已有同源基因在玉米里被报道响应干旱胁迫和盐胁迫[31,51]。另外荧光定量PCR实验也证明这些核心基因对其模块对应的非生物胁迫响应, 故推测这些调节酶、特殊蛋白以及转录因子位于玉米响应非生物胁迫的调控中心, 可能在玉米生长发育及非生物胁迫防御反应等过程中具有重要作用。
4 结论
本研究利用WGCNA对低温胁迫、高温胁迫、干旱胁迫和盐胁迫4种非生物胁迫相关基因做共表达网络分析, 得到25个模块, 确定了4个分别响应4种非生物胁迫的模块和同时响应4种胁迫的模块。对这5个模块作基因互作网络分析, 预测了模块中10个核心基因, 这10个基因可能为非生物胁迫模块的核心基因。本研究通过大数据网络分析, 为玉米抗逆基因的克隆和非生物胁迫调控途径和机制研究提供重要的参考依据。
附表 请见网络版: 1) 本刊网站http://zwxb.chinacrops. org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodicalzuowxb.aspx。
[1] Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis., 2005, 4: 17.
[2] Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis., 2008, 9: 559.
[3] Kuang J F, Wu C J, Guo Y F, Walther D, Shan W, Chen J Y, Chen L, Lu W J. Deciphering transcriptional regulators of banana fruit ripening by regulatory network analysis., 2021;19: 477–489.
[4] Sun S, Xiong X P, Zhu Q, Li Y J, Sun J. Transcriptome sequencing and metabolome analysis reveal genes involved in pigmentation of green-colored cotton fibers., 2019, 20: 4838.
[5] Greenham K, Guadagno C R, Gehan M A, Mockler T C, Weinig C, Ewers B E, McClung C R. Temporal network analysis identifies early physiological and transcriptomic indicators of mild drought in., 2017, 6: e29655.
[6] 马娟, 曹言勇, 王利锋, 李晶晶, 王浩, 范艳萍, 李会勇. 利用WGCNA鉴定玉米株高和穗位高基因共表达模块. 作物学报, 2020, 46: 385–394.
Ma J, Cao Y Y, Wang L F, Li J J, Wang H, Fan Y P, Li H Y. Identification of gene co-expression modules of maize plant height and ear height by WGCNA., 2020, 46: 385–394 (in Chinese with English abstract)
[7] Larcher W. Physiological Plant Ecology. England: J R Etherington, 1996. pp 630–631.
[8] Krasensky J, Jonak C. Drought, salt, and temperature stress- induced metabolic rearrangements and regulatory networks., 2012, 63: 1593–1608
[9] Richards R A. Defining selection criteria to improve yield under drought., 1996, 20: 157–166.
[10] Cushman J C, Bohnert H J. Genomic approaches to plant stress tolerance., 2000, 3: 117–124
[11] Li M, Sui N, Lin L, Yang Z, Zhang Y. Transcriptomic profiling revealed genes involved in response to cold stress in maize., 2019, 46: 830–844.
[12] Frey F P, Pitz M, Schön C C, Hochholdinger F. Transcriptomic diversity in seedling roots of European flint maize in response to cold., 2020, 21: 300–310.
[13] Guo J, Li C, Zhang X, Li Y, Zhang D, Shi Y, Song Y, Li Y, Yang D, Wang T. Transcriptome and GWAS analyses reveal candidate gene for seminal root length of maize seedlings under drought stress., 2020, 292: 110380.
[14] Cao L, Lu X, Wang G, Zhang P, Fu J, Wang Z, Wei L, Wang T. Transcriptional regulatory networks in response to drought stress and rewatering in maize (L.)., 2021, 6: 1203–1219.
[15] Li W Z, Hao Z F, Pang J L, Zhang M, Wang N, Li X H, Li W H, Wang L, Xu M Y. Effect of water-deficit on tassel development in maize., 2019, 681: 86–92.
[16] Wang H Q, Liu P, Zhang J W, Zhao B, Ren B Z. Endogenous hormones inhibit differentiation of young ears in maize (L.) under heat stress., 2020, 11: 533040.
[17] Waters A J, Makarevitch I, Noshay J, Burghardt L T, Hirsch C N, Hirsch C D, Springer N M. Natural variation for gene expression responses to abiotic stress in maize., 2017, 89: 706–717.
[18] Wang M Q, Wang Y F, Zhang Y F, Li C X, Gong S C, Yan S Q, Li G L, Hu G H, Ren H L, Yang J F, Yu T, Yang K J. Comparative transcriptome analysis of salt-sensitive and salt-tolerant maize reveals potential mechanisms to enhance salt resistance., 2019, 41: 781–801.
[19] Goldberg D H, Victor J D, Gardner E P, Gardner D. Spike train analysis toolkit: enabling wider application of information- theoretic techniques to neurophysiology., 2009, 7: 165–178.
[20] Kroll K W, Mokaram N E, Pelletier A R, Frankhouser D E, Westphal M S, Bundschuh R, Blachly J S, Yan P. Quality control for RNA-Seq (QuaCRS): an integrated quality control pipeline., 2014, 13: 7–14.
[21] Bolger A M, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data., 2014, 30: 2114–2120.
[22] Kim D, Langmead B, Salzberg S L. HISAT: a fast spliced aligner with low memory requirements., 2015, 12: 357–360.
[23] Liao Y, Smyth G K, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features., 2014, 30: 923–930.
[24] Ghosh S, Chan C K. Analysis of RNA-Seq data using TopHat and Cufflinks., 2016, 1374: 339–361.
[25] Tian T, Liu Y, Yan H Y, You Q, Yi X, Du Z, Xu W Y, Su Z. agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update., 2017,45: W122–W129.
[26] Su G, Morris J H, Demchak B, Bader G D. Biological network exploration with Cytoscape 3., 2014, 47: 8.13.1–8.13.24.
[27] Zeng R, Li Z Y, Shi Y T, Fu D, Yin P, Cheng J K, Jiang C F, Yang S H. Natural variation in a type-A response regulator confers maize chilling tolerance., 2021, 12: 4713.
[28] Wang X L, Wang H W, Liu S X, Ferjani A, Li J S, Yan J B, Yang X H, Qin F. Genetic variation incontributes to drought tolerance in maize seedlings., 2016, 48: 1233–1241.
[29] Pan Z Y, Liu M, Zhao H L, Tan Z D, Liang K, Sun Q, Gong D M, He H J, Zhou W Q, Qiu F Z.is involved in drought tolerance by maintaining cuticular wax structure in maize., 2020, 62: 1895–1909.
[30] Li X D, Gao Y Q, Wu W H, Chen L M, Wang Y. Two calcium-dependent protein kinases enhance maize drought tolerance by activating anion channelin guard cells., 2022, 20: 143–157.
[31] Xiang Y L, Sun X P, Gao S, Qin F, Dai M Q. Deletion of an endoplasmic reticulum stress response element in a ZmPP2C-A gene facilitates drought tolerance of maize seedlings., 2017, 10: 456–469.
[32] Mao H D, Wang H W, Liu S X, Li Z Q, Yang X H, Yan J B, Li J S, Tran L S P, Qin F. A transposable element in a NAC gene is associated with drought tolerance in maize seedlings., 2015, 6: 8326.
[33] Li L, Du Y C, He C, Dietrich C R, Li J K, Ma X L, Wang R, Liu Q, Liu S Z, Wang G Y, Schnable P S, Zheng J. Maize glossy6 is involved in cuticular wax deposition and drought tolerance., 2019, 70: 3089–3099.
[34] Vaughan M M, Christensen S, Schmelz E A, Huffaker A, McAuslane H J, Alborn H T, Romero M, Allen L H, Teal P E. Accumulation of terpenoid phytoalexins in maize roots is associated with drought tolerance., 2015, 38: 2195–2207.
[35] Zhu D, Chang Y, Pei T, Zhang X L, Liu L, Li Y, Zhuang J H, Yang H L, Qin F, Song C P, Ren D T. MAPK-like protein 1 positively regulates maize seedling drought sensitivity by suppressing ABA biosynthesis., 2020, 102: 747–760.
[36] Zhu J T, Wang G L, Li C L, Li Q Q, Gao Y K, Chen F G, Xia G M. Maize Sep15-like functions in endoplasmic reticulum and reactive oxygen species homeostasis to promote salt and osmotic stress resistance., 2019, 42: 1486–1502.
[37] Lund A A, Blum P H, Bhattramakki D, Elthon T E. Heat-stress response of maize mitochondria., 1998, 116: 1097–1110.
[38] Li Z X, Srivastava R, Tang J, Zheng Z H, Howell S H.-effects condition the induction of a major unfolded protein response factor,, in response to heat stress in maize., 2018, 9: 833.
[39] Zhang N, Huang X Q. Mapping quantitative trait loci and predicting candidate genes for leaf angle in maize., 2021, 16: e0245129.
[40] Li Z X, Howell S H. Heat stress responses and thermotolerance in maize., 2021, 22: 948.
[41] Holubová Ľ, Švubová R, Slováková Ľ, Bokor B, Kročková V C, Renčko J, Uhrin F, Medvecká V, Zahoranová A, Gálová E. Cold atmospheric pressure plasma treatment of maize grains-induction of growth, enzyme activities and heat shock proteins., 2021, 22: 8509.
[42] Jiang Y L, Zheng Q Q, Chen L, Liang Y N, Wu J D. Ectopic overexpression of maize heat shock transcription factor geneconfers increased thermos- and salt-stress tolerance in transgenic., 2018, 40: 9.
[43] Jiménez-González A S, Fernández N, Martínez-Salas E, Sánchez de Jiménez E. Functional and structural analysis of maize hsp101 IRES., 2014, 9: e107459.
[44] Luo X, Wang B C, Gao S, Zhang F, Terzaghi W, Dai M Q. Genome-wide association study dissects the genetic bases of salt tolerance in maize seedlings., 2019, 61: 658–674.
[45] Augustine R C, York S L, Rytz T C, Vierstra R D. Defining the SUMO system in maize: SUMOylation is up-regulated during endosperm development and rapidly induced by stress., 2016, 171: 2191–2210.
[46] Gu L K, Liu Y K, Zong X J, Liu L, Li D P, Li D Q. Overexpression of maize mitogen-activated protein kinase gene,inincreases tolerance to salt stress., 2010, 37: 4067–4073.
[47] Li H Y, Du H M, Huang K F, Chen X, Liu T Y, Gao S B, Liu H L, Tang Q L, Rong T Z, Zhang S Z. Identification, and functional and expression analyses of the CorA/MRS2/MGT-Type magnesium transporter family in maize., 2016, 57: 1153–1168.
[48] Zhu J T, Wang G L, Li C L, Li Q Q, Gao Y K, Chen F G, Xia G M. Maize Sep15-like functions in endoplasmic reticulum and reactive oxygen species homeostasis to promote salt and osmotic stress resistance., 2019, 42: 1486–1502.
[49] Ma H Z, Liu C, Li Z X, Ran Q J, Xie G N, Wang B M, Fang S, Chu J F, Zhang J R. ZmbZIP4 contributes to stress resistance in maize by regulating ABA synthesis and root development., 2018, 178: 753–770.
[50] Kong M S, Luo M J, Li J N, Feng Z, Zhang Y X, Song W, Zhang R Y, Wang R H, Wang Y D, Zhao J R, Tao Y S, Zhao Y X. Genome-wide identification, characterization, and expression analysis of the monovalent cation-proton antiporter superfamily in maize, and functional analysis of its role in salt tolerance., 2021, 113: 1940–1951.
[51] Cao Y B, Zhang M, Liang X Y, Li F R, Shi Y L, Yang X H, Jiang C F. Natural variation of an EF-hand Ca2+-binding-protein coding gene confers saline-alkaline tolerance in maize., 2020, 11: 186.
[52] Fang X, Li W, Yuan H T, Chen H W, Bo C, Ma Q, Cai R H. Mutation of ZmWRKY86 confers enhanced salt stress tolerance in maize., 2021, 67: 840–850.
[53] Luo J, Yu C M, Yan M, Chem Y H. Molecular characterization of the promoter of the stress-induciblegene in maize., 2020, 64: 200–210.
[54] Lin M, Matschi S, Vasquez M, Chamness J, Kaczmar N, Baseggio M, Miller M, Stewart E L, Qiao P F, Scanlon M J, Molina I, Smith L G, Gore M A. Genome-wide association study for maize leaf cuticular conductance identifies candidate genes involved in the regulation of cuticle development.(Bethesda), 2020, 10: 1671–1683.
[55] Ge C X, Wang Y G, Lu S, Zhao X Y, Hou B K, Balint-Kurti P J, Wang G F. Multi-omics analyses reveal the regulatory network and the function of ZmUGTs in maize defense response., 2021, 12: 738261.
[56] Sallam N, Moussa M. DNA methylation changes stimulated by drought stress in ABA-deficient maize mutant., 2021, 160: 218–224.
[57] Shao H B, Chu L Y, Shao M A, Jaleel C A, Mi H M. Higher plant antioxidants and redox signaling under environmental stresses., 2008, 331: 433–441.
[58] Wang J F, Shen Q R. Roles of organic acid metabolism in plant adaptation to nutrient deficiency and aluminum toxicity stress., 2006, 17: 2210–2216.
[59] Raina M, Kisku A V, Joon S, Kumar S, Kumar D. Calcium Transport Elements in Plants. The United States of America: Academic Press, 2021. pp 231–248.
[60] Shigeoka S, Maruta T. Cellular redox regulation, signaling, and stress response in plants., 2014, 78: 1457–1470.
[61] Jang J C. Arginine-rich motif-tandem CCCH zinc finger proteins in plant stress responses and post-transcriptional regulation of gene expression., 2016, 252: 118–124.
[62] Wu X W, Bacic A, Johnson K L, Humphries J. The role of brachypodium distachyon Wall-Associated Kinases (WAKs) in cell expansion and stress responses., 2020, 9: 2478.
Identification of abiotic stress-related gene co-expression networks in maize by WGCNA
DENG Zhao**, JIANG Huan-Qi**, CHENG Li-Sha, LIU Rui, HUANG Min, LI Man-Fei*, and DU He-Wei*
College of Life Sciences, Yangtze University, Jingzhou 434025, Hubei, China
Weighted Gene Co-expression Network Analysis (WGCNA) is a classic systems biology analysis method, which can be used to identify coexpressed gene modules and explore the biological correlation between modules and target traits, and mine core genes in module networks. In this study, 58 transcriptome data of roots, stems, leaves, and other tissues under low temperature stress, high temperature stress, drought stress, and salt stress in maize (L.) were collected, and the gene co-expression network of maize abiotic stress was identified by WGCNA method. After filtering the 12,552 low-expression genes from transcriptome data, the co-expression network was constructed using the remaining 27,204 high-expression genes, and 25 modules were obtained. According to the distribution of abiotic stress-related genes and different expression genes in the modules reported in maize, the mediumpurple4, ivory, coral2, darkseagreen4 modules most related to low temperature stress, high temperature stress, drought and salt stresses, and green modules responding to various stresses were screened out. Subsequently, GO enrichment of the genes in these five modules revealed that genes with functions related to abiotic stress were significantly enriched in these modules, such as stress response, peroxidase activity. Correlation analysis showed that 10 abiotic stress-related core genes were predicted, including,,,, and. This study provides new ideas for the mining of abiotic stress-related genes and the research of abiotic stress regulatory networks in maize.
maize; abiotic stress; WGCNA; core gene
10.3724/SP.J.1006.2023.23017
本研究由国家自然科学基金项目(31771801,32072069),湖北省高等学校优秀中青年创新团队项目(2017T04)和湖北省大学生创新创业项目(Yz2021220)资助。
This study was supported by the National Natural Science Foundation of China (31771801, 32072069), the Outstanding Young and Middle-aged Innovation Team Project of Hubei Province (2017T04), and the College Students’ Innovation and Entrepreneurship Training Program of Hubei Province (Yz2021220).
通信作者(Corresponding authors):李曼菲, E-mail: mfli_maize@163.com; 杜何为, E-mail: duhewei666@163.com
同等贡献(Contributed equally to this work)
邓照, E-mail: 282175698@qq.com
2022-02-22;
2022-06-07;
2022-07-08.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20220707.1352.010.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
猜你喜欢
真空与低温(2022年6期)2023-01-06 07:33:20
今日农业(2022年16期)2022-11-09 23:18:44
今日农业(2022年15期)2022-09-20 06:55:48
环球时报(2022-06-20)2022-06-20 17:06:23
现代塑料加工应用(2021年5期)2021-02-28 08:18:04
昆明医科大学学报(2020年12期)2021-01-26 00:44:02
基层中医药(2018年8期)2018-11-10 05:32:06
劳动保护(2018年8期)2018-09-12 01:16:18
山东医药(2017年20期)2017-07-01 19:52:07
中国学术期刊文摘(2016年1期)2016-02-13 14:05:23
