
玉米株高和穗位高性状全基因组关联分析
2023-01-16 08:36马雅杰鲍建喜高悦欣李雅楠秦文萱王彦博李金萍董振营万向元
作物学报 2023年3期
马雅杰 鲍建喜 高悦欣 李雅楠 秦文萱 王彦博 龙 艳 李金萍 董振营* 万向元*
玉米株高和穗位高性状全基因组关联分析
马雅杰1,**鲍建喜1,**高悦欣1李雅楠1秦文萱1王彦博1龙 艳1李金萍2董振营1,2,*万向元1,2,*
1北京科技大学生物与农业研究中心 / 化学与生物工程学院 / 顺德研究生院 / 北京中智生物农业国际研究院, 北京 100083;2北京首佳利华科技有限公司 / 主要作物生物育种北京市工程实验室 / 生物育种北京市国际科技合作基地, 北京 100192
适宜的株高和穗位高可提高植株的养分利用效率及抗倒伏性, 对玉米增产和稳产具有重要意义。为揭示玉米株高和穗位高遗传机制, 本研究以854份玉米自交系为关联群体, 利用均匀分布于玉米10条染色体的2795个SNP标记对4个环境下玉米株高、穗位高以及穗位系数进行全基因组关联分析(genome-wide association study, GWAS)。共定位到81个显著关联SNP位点(<0.0001), 其中与株高显著关联的SNP为35个, 单个位点表型解释率为0.02%~6.23%; 与穗位高显著关联SNP为31个, 单个位点表型变异解释率为0.03%~3.06%; 与穗位系数显著关联的SNP位点为24个, 单个位点表型变异解释率为0.03%~6.64%。进一步鉴定出15个可在2个及以上环境共定位的稳定SNP, 其中6个为本研究首次发现, 9个位于前人定位QTL区间或/和关联SNP位点2 Mb范围内。在15个稳定SNP位点上下游各200 kb的置信区间共发现83个功能注释基因, 结合文献分析筛选出了每个位点最有可能的候选基因, 这些候选基因主要参与激素合成与信号转导、糖类代谢、细胞分裂调控等途径。鉴定出6个主效SNP位点, 并发现1个可同时调控株高、穗位高和穗位系数的一因多效位点。本研究可为分子标记辅助选择育种提供有效遗传位点, 为精细定位和克隆株高与穗位高相关性状基因提供参考。
玉米; 株高; 穗位高; 全基因组关联分析; 候选基因
玉米是世界重要粮食作物, 同时大量用作动物饲料和工业原料, 在社会生产和生活中具有重要地位[1-3]。据国家统计局(http://www.stats.gov.cn/)数据, 我国玉米播种面积在2020年达到4126万公顷, 为我国种植面积最大的农作物。因此, 提高玉米单产对保障我国粮食安全具有重要意义[4-5]。适宜的株高(plant height, PH)和穗位高(ear height, EH)可提高植株的养分利用效率、改善株型结构, 进而有利于提高单产[6-9]。同时, 相对较低的株高和穗位高有利于降低玉米植株重心, 提高抗倒伏性, 从而有利于玉米稳产[10-13]。
玉米株高、穗位高均属于典型数量性状, 目前国内外已有通过QTL (quantitative trait locus)定位以及全基因组关联分析(genome-wide association analysis, GWAS)等方法挖掘株高及穗位高等性状遗传位点的研究报道。对于株高性状, Beavis等[14]较早通过构建多个F2群体, 利用RFLP标记定位到14个株高QTL, 分析结果显示其中大多数QTL与已克隆株高调控基因位点吻合。Yan等[15]通过创制F2:3群体, 利用RFLP及SSR标记构建遗传连锁图谱, 在玉米发育的5个不同时期共定位8个株高QTL, 其中3个QTL可在不同发育时期均被检测到, 表明这些QTL在玉米不同发育时期均对株高具有调控作用。Weng等[16]利用包含44,235个有效SNP位点的MaizeSNP50芯片, 基于混合线性模型(mixed linear model, MLM)对284个玉米自交系进行株高性状GWAS分析, 共检测到204个显著关联SNP位点, 定位区间发现赤霉素、生长素和表观遗传途径相关基因, 暗示这些基因可能参与了优良自交系矮化表型形成。对于穗位高性状, Bai等[17]利用以Zong3为背景的近等基因导入系和回交群体以及120个SSR标记进行QTL定位, 共检测到15个穗位高QTL, 其单个QTL表型变异解释率为5.6%~31.2%, 同时检测到3个在不同年份间较为稳定的位点。Vanous等[18]利用包含252个双单倍体株系的关联群体和采用GBS (genotyping-by-sequencing)方法所获得的62,077个SNP标记进行GWAS分析, 共鉴定出30个显著SNP位点以及20个与生长素和赤霉素信号等途径相关的候选基因。针对穗位系数(EH/PH), Wang等[19]通过构建1021个重组自交系和采用GBS方法所获得的16,769个SNP标记, 共定位到8个QTL, 其中一些QTL区间包含已知可调控茎秆伸长的基因如、、和。刘坤等[20]以284份自交系为关联群体, 利用56万个SNP标记进行GWAS分析, 共关联到5个与穗位系数性状显著关联的位点, 其中位于8号染色体的位点可解释10.12%的表型变异。
1 材料与方法
1.1 试验材料
以854份玉米自交系为供试群体材料, 将该群体分别在2020年和2021年种植于北京平谷(PG)和山东诸城(ZC) 4个环境, 即20PG、20ZC、21PG和21ZC。每材料双行区种植, 行长6.0 m, 行距0.6 m, 株距0.2 m, 其他同常规大田管理。
1.2 表型测定与分析
在玉米乳熟期, 每个家系随机选取5个单株, 测量株高(PH)和穗位高(EH)。株高指地面至雄穗顶端的距离, 穗位高指地面至第1果穗着生节的距离, 单位均为cm, 穗位系数(EH/PH)为穗位高和株高的比值。采用IBM SPSS Statistics 23.0软件对3类性状均值、标准差、变异系数、变异范围、偏度、峰度进行描述性统计分析。参照Knapp等[21]方法计算广义遗传力2,2g2g2gl2gy2e2。式中,2表示广义遗传力,g2为遗传方差,gl2为基因型与地点互作方差,gy2为基因型与年份互作方差,e2为残差方差,为地点数,为年份数。利用Origin 2018软件计算不同性状各环境间的皮尔森相关系数(Pearson correlation coefficient), 利用R包“lme4”计算株高、穗位高和穗位系数的最佳线性无偏预测值(best linear unbiased prediction, BLUP)。
1.3 DNA提取与基因型检测
在玉米五叶期, 取关联群体植株的叶片, CTAB法[22]提取DNA, 采用由北京市农林科学院玉米研究中心研发的包含3072个SNP位点的MaizeSNP3072芯片[23]进行基因分型。去除杂合率大于10%、最小等位基因频率小于5%和缺失大于20%的SNP, 最终筛选出2795个高质量的SNP标记, 利用Structure 2.3.4软件进行群体结构分析[24], 利用FastTree软件邻接法(Neighbor-Joining, NJ)构建关联分析群体系统进化树。
1.4 全基因组关联分析
利用R包“FarmCPU”进行全基因组关联分析[25], 以群体结构-主成分(principal components analysis, PCA)为协变量。Bonferroni校正作为最严格的多重假设检验校正方法, 可能导致较高的假阴性[26-27], 根据Bonferroni校正,= 0.05/2795 = 1.79E-05 (即–log10() = 4.75)。为减少假阴性, 本研究取–log10() = 4 (即< 1E-04)为显著性阈值[16,28-30]。利用线性回归方法[31]计算表型变异解释率(phenotypic variation explained, PVE)。利用R包“CMplot”绘制曼哈顿图及QQ-PLOT图。
1.5 候选基因功能注释
利用Tassel 5.0软件计算群体LD (linkage disequilibrium)衰减距离[32], 结合Maize GDB (https:// www.maizegdb.org/)数据库B73 (RefGen_v4)参考基因组确定区间内候选基因, 基于UniProt数据库(http://www.uniprot.org/)和NCBI数据库(NCBI, http://www.ncbi.nlm.nih.gov/)基因功能注释和对比文献分析筛选出最可能的候选基因[33]。
2 结果与分析
2.1 表型统计分析
对于株高性状, 4个环境下均值在179.73~209.99 cm之间, 表型变异范围在79.00~291.00 cm之间, 变异系数在13.05%~13.40%; 对于穗位高性状, 4个环境下均值在58.91~79.53 cm之间, 表型变异范围在19.50~139.40 cm之间, 变异系数在19.90%~22.24%; 对于穗位系数性状, 4个环境下均值在0.33~0.39之间, 表型变异范围在0.18~0.59之间, 变异系数在12.82%~15.79% (表1)。3个表型性状在各个环境变异系数均大于10%, 表明该群体中所研究农艺性状存在较为丰富的表型变异。3个性状偏度和峰度绝对值均小于1, 数据分布曲线符合正态分布(图1), 说明株高、穗位高以及穗位系数符合数量性状特征。

(图1)
处理缩写同表1。Abbreviations are the same as those given in Table 1.
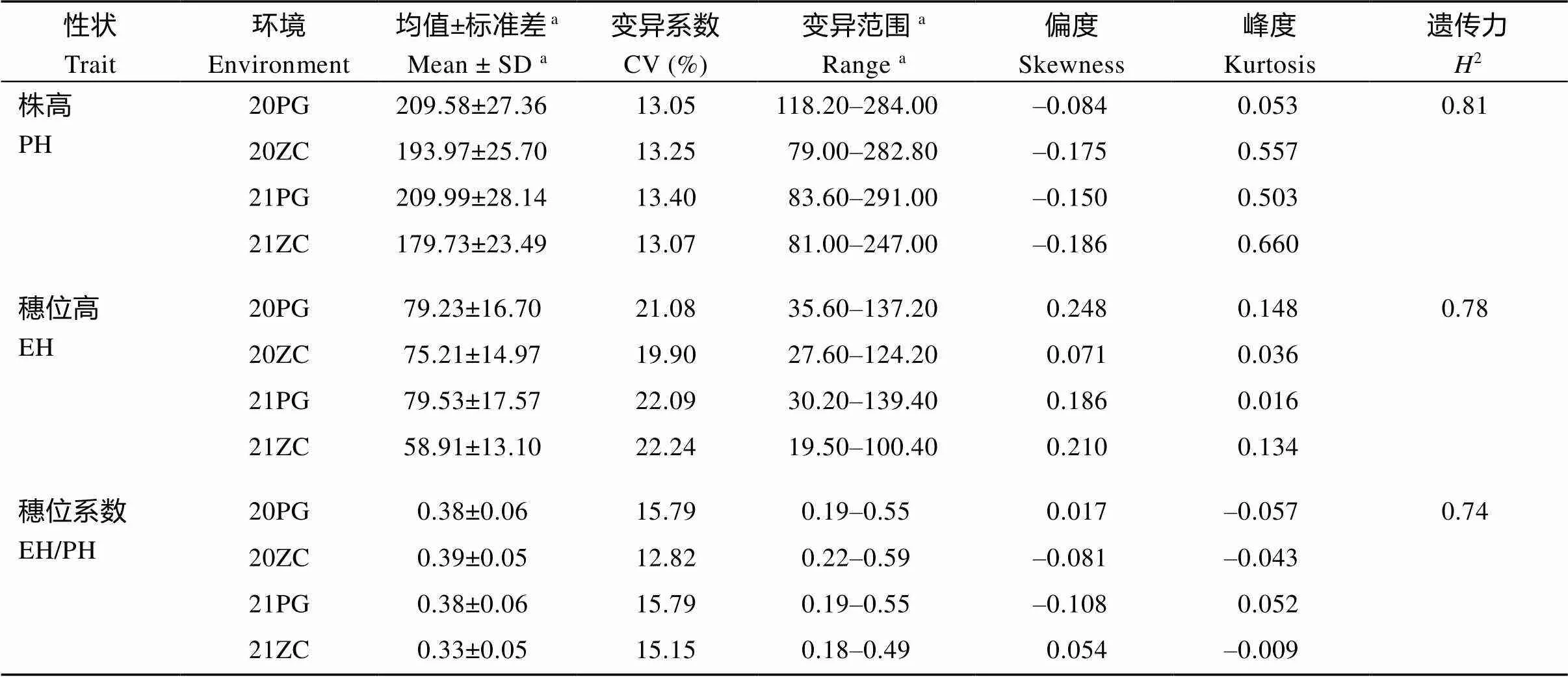
表1 株高、穗位高和穗位系数表型数据统计分析
a: 株高和穗位高单位为cm。PH、EH和EH/PH分别表示株高、穗位高和穗位系数; PG、ZC分别表示平谷和诸城。
a: the units of plant height and ear height are in cm. PH, EH, and EH/PH represent plant height, ear height, and the ratio of EH to PH, respectively; PG and ZCrepresent Pinggu and Zhucheng, respectively.
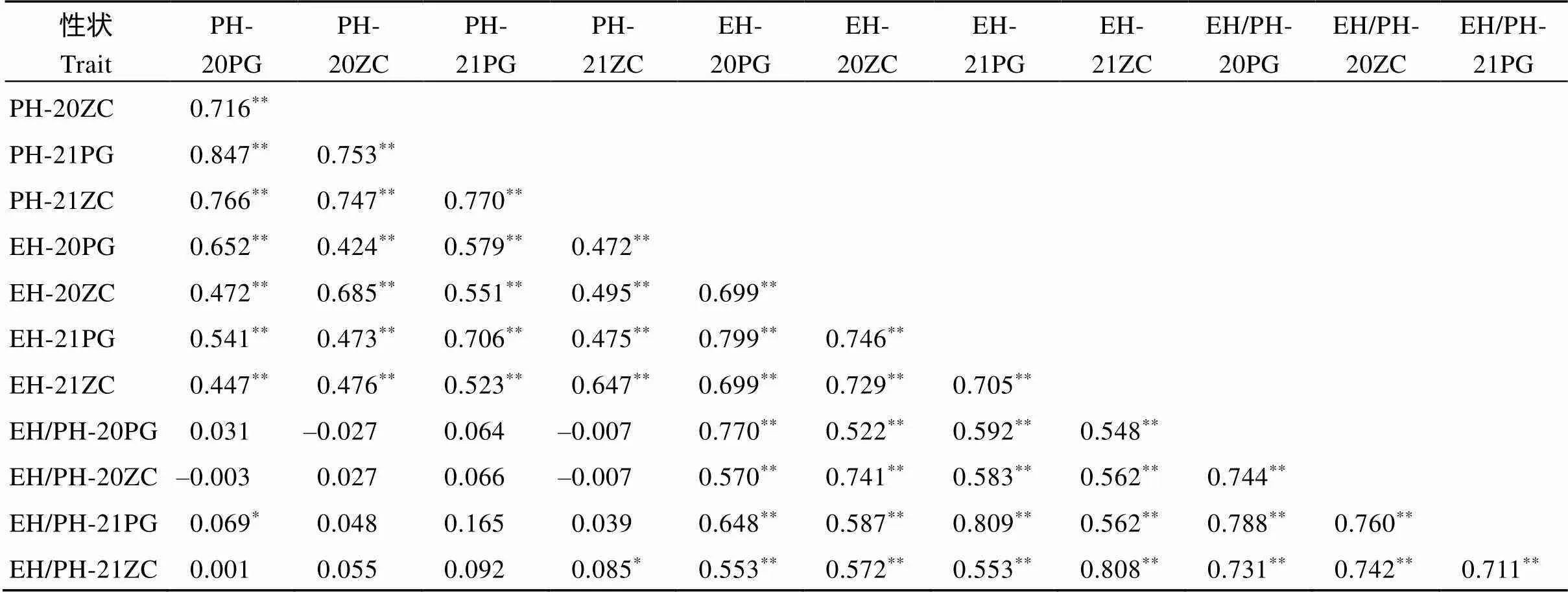
表2 不同环境间PH、EH和EH/PH性状相关性分析
*: 在< 0.05水平显著相关;**在< 0.01水平显著相关。处理缩写同表1。
*:< 0.05;**:< 0.01. Abbreviations are the same as those given in Table 1.
相关性分析表明不同环境间同一性状数据均呈极显著相关(< 0.01), 株高、穗位高和穗位系数性状相应相关系数分别为0.716~0.847、0.699~0.799和0.711~0.788 (表2)。不同性状之间相关性分析表明株高与穗位高性状之间呈极显著正相关(< 0.01),相关系数在0.424~0.706之间; 穗位高与穗位系数之间同样呈极显著正相关(< 0.01), 相关系数在0.522~0.809之间; 而株高与穗位系数之间只在少数环境下呈微弱正相关(表2)。株高广义遗传力为0.81, 穗位高广义遗传力为0.78, 穗位系数广义遗传力为0.74, 表明株高、穗位高以及穗位系数主要受遗传因素影响(表1), 可以进行后续分析。
2.2 全基因组关联分析
利用2795个高质量SNP分析854份玉米自交系遗传多样性, 发现本群体材料大致分为10个类群, 表明具有较高遗传多样性(附图1)。在考虑群体结构情况下利用“FarmCPU”模型对4个环境株高、穗位高以及穗位系数表型值及其BLUP育种值分别进行全基因组关联分析。共检测到81个显著关联SNP位点, 其中与株高性状显著关联的SNP位点有35个, 单个SNP位点表型变异解释率范围为0.02%~ 6.23%; 与穗位高显著关联的SNP位点有31个, 单个SNP位点表型变异解释率范围为0.03%~3.06%; 与穗位系数显著关联的SNP位点有24个, 单个SNP位点表型变异解释率范围为0.03%~6.64% (附表1)。进一步发现株高与穗位高性状共定位SNP有5个, 穗位高与穗位系数性状共定位SNP有4个, 而株高与穗位系数性状未发现共定位SNP位点, 这与表型相关性分析中株高与穗位系数基本无显著相关的结果相吻合(表2和附表1)。将每个性状中被检测到2次及以上的SNP视为稳定SNP位点, 鉴定出8个与株高性状显著相关的稳定SNP位点, 分别位于1号、2号、4号、5号染色体, 其中值最低的SNP位点为PZE-104102768 (5.59E-06), PVE最高的SNP位点为PZE-101256370 (5.19%), 被检测到次数最多位点为PZE-105102182, 分别在BLUP和20ZC、21PG、21ZC环境中被检测到(表3)。与穗位高性状显著关联的稳定SNP位点为4个, 分别位于1号、5号、8号染色体, P 值最低位点为PZE-108069615 (3.04E-07), PVE 最高位点为PZE-105098019 (3.00%) (表3)。穗位系数性状关联分析中检测到4个稳定SNP 位点, 分别位于3号、8号染色体, P 值最低SNP 位点为PZE-103083718(2.27E-06), PVE 最高SNP 位点为PZE-108069615(6.64%) (表3)。鉴定出1个位于8号染色体穗位高与穗位系数共定位稳定SNP 位点PZE-108069615, 该位点可解释2.36%穗位高性状表型变异和6.64%穗位系数性状表型变异(表3)。
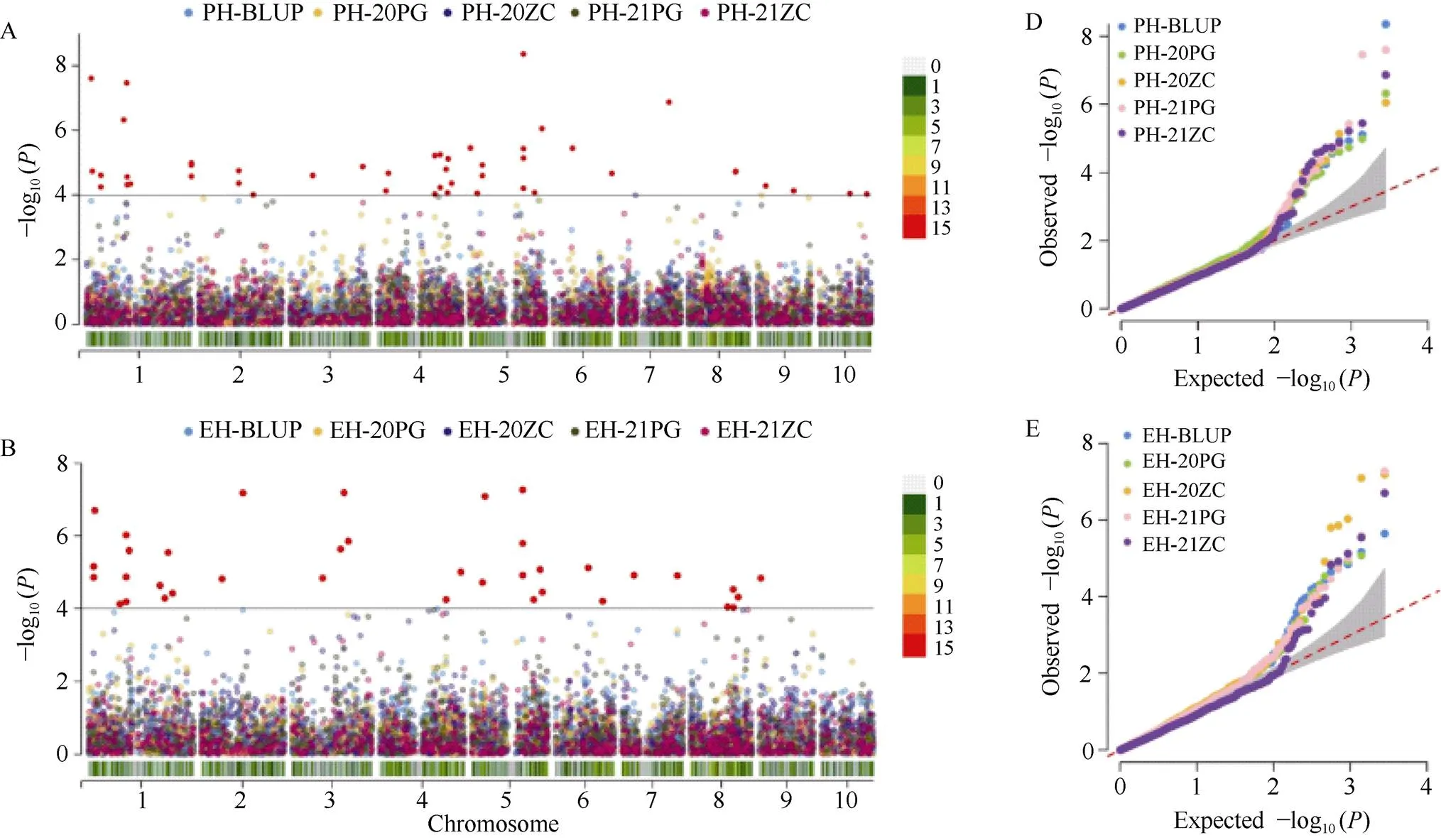
(图2)
A、B、C分别为株高、穗位高、穗位系数性状关联分析的曼哈顿图; D、E、F分别为株高、穗位高、穗位系数性状关联分析的QQ图。BLUP表示最佳线性无偏预测值。其他处理缩写同表1。
A, B, and C were Manhattan plots for GWAS of PH, EH, and EH/PH, respectively. D, E, and F were QQ plots for GWAS of PH, EH, and EH/PH, respectively. BLUP represents best linear unbiased prediction. Other abbreviations are the same as those given in Table 1.
2.3 等位变异效应分析
分别选取株高、穗位高和穗位系数显著相关稳定SNP位点中PVE最大的2个SNP作为主效SNP, 并分析其等位变异效应。对于株高性状, 发现2个主效SNP不同等位变异在20PG、20ZC、21PG和21ZC 4个环境均可导致极显著表型差异, 如PZE-101109358位点的G/G等位变异在4个环境下的平均株高比A/A等位变异降低8.81~10.81 cm, PZE-101256370位点的A/A等位变异在4个环境下的平均株高比G/G等位变异降低8.74~11.51 cm (图3-A)。对于穗位高性状, 携带2个主效SNP不同等位变异的自交系材料在4个环境下表型同样呈显著差异。其中PZE-105098019位点的G/G等位变异在4个环境下平均穗位高比A/A等位变异降低4.24~7.81 cm; PZE-101024808位点的C/C等位变异在4个环境下的平均穗位高比A/A等位变异降低2.13~6.06 cm (图3-A)。对于穗位系数性状, PZE-103075978位点的G/G等位变异在4个环境下导致平均穗位系数比A/A等位变异降低0.020~0.031, PZE-108069615位点的G/G等位变异则比A/A等位变异平均穗位系数降低0.017~0.032 (图3-A)。
21世纪已经成为名副其实的互联网时代,互联网成为创新的典型代表,也反过来极大促进了创新创业工作。近年来,互联网逐渐从“网络化”发展为“智能化”,这使得网络思想政治教育一直处于追踪和把握新技术的过程。从图4可以看出,以报纸、杂志、电视、广播为代表的传统大众传媒已日趋势微,而以门户网站和“两微一端”为代表的互联网和新媒体平台成为了青年学生了解时事热点的最重要渠道。
在稳定SNP位点中, 株高性状关联位点PZE-105102182被重复检测到的次数最多, 推测该位点携带可在不同环境稳定调控株高性状的重要基因, 因此对该位点进一步分析。发现A/A等位变异导致株高性状在20PG、20ZC、21PG和21ZC环境平均比G/G等位变异分别降低4.51、7.04、5.48和4.64 cm (图3-B)。进一步分析穗位高和穗位系数性状, 发现A/A等位变异导致20PG、20ZC、21PG和21ZC环境的平均穗位高比G/G等位变异分别降低5.42、7.01、4.91和5.02 cm, 穗位系数则分别降低0.018、0.022、0.016、0.019, 差异均达到极显著水平(图3-B), 表明PZE-105102182位点具有一因多效性, A/A为优异等位变异, 可同时负调控株高、穗位高和穗位系数性状。
2.4 候选基因分析
以2降至0.2所对应物理距离为该群体LD衰减距离[34-35], 计算得到该群体的衰减距离约为200 kb, 与前人结果较为一致[36-38]。在3个性状检测到的15个稳定SNP位点上下游200 kb置信区间共发现173个候选基因, 其中具有功能注释基因为83个。通过前期总结, 发现玉米分生组织调控、微管活动、蔗糖运输、根毛生长、光敏色素合成相关基因以及激素信号、AP2类转录因子等的相关基因可调控玉米株高、穗位高性状[33]。因此, 若置信区间存在以上生物途径相关基因, 则筛选显著SNP标记最近的该类基因作为候选基因; 若置信区间无以上生物途径相关基因, 则以距离显著SNP标记最近、且具有功能注释的基因为候选基因。共筛选到15个候选基因, 进一步分析发现6个候选基因与激素的合成与信号转导途径相关, 分别为。5个候选基因与糖类代谢相关, 分别为; 3个候选基因参与细胞分裂调控途径, 分别为。针对特定性状, 2个株高性状主效位点候选基因分别为编码脱落酸(ABA)信号转导途径核质转运蛋白的Zm00001d034914和编码甘露糖苷酶的。2个穗位高性状主效位点候选基因分别为编码RING/U-box泛素连接酶的和编码岩藻糖基转移酶蛋白的。2个穗位系数性状主效位点候选基因分别为编码CPP转录因子的和编码Dof锌指蛋白的。
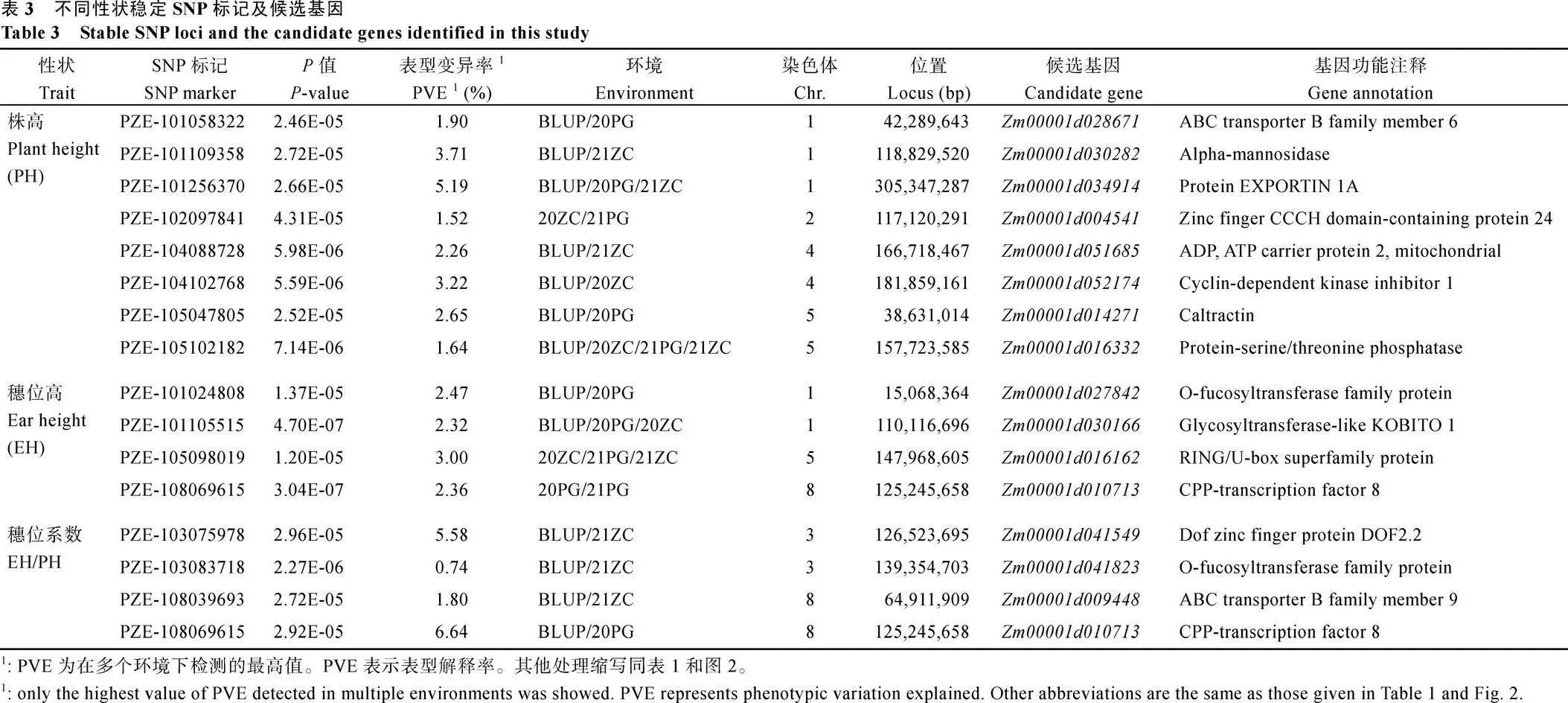
图3 株高、穗位高和穗位系数相关重要SNP位点等位变异效应分析
Fig. 3 Allelic effects of the SNPs associated with plant height (PH), ear height (EH), and the ratio of EH/PH (EH/PH) traits
A: 株高、穗位高、穗位系数主效SNP位点等位变异效应分析; B: PZE-105102182位点对不同性状的等位效应分析。*: 在< 0.05水平上显著相关;**: 在< 0.01水平上显著相关。处理缩写同表1。
A: the allelic effect of dominant SNP locus in plant height, ear height, and the ratio of EH to PH; B: the allelic effect of PZE-105102182 locus on different traits.*: significance correlation at< 0.05;**: significant correlation at< 0.01. Abbreviations are the same as those given in Table 1.
3 讨论
3.1 株高、穗位高相关性状定位结果分析
株高、穗位高及穗位系数, 为多基因控制的数量性状, 其直接影响植株的养分利用率及抗倒伏性[14-20]。全基因组关联分析方法具有高分辨率和高通量的优势, 可以高效鉴定调控重要性状关键位点和其优异等位变异[39]。本研究利用854份自交系群体进行株高、穗位高及穗位系数性状GWAS, 共检测到81个显著关联SNP位点, 株高与穗位高、穗位高与穗位系数均鉴定出共定位SNP, 未发现株高与穗位系数性状共定位位点。可见穗位高和株高性状可能存在相似调控机制, 但是穗位系数与株高调控机制差异较大。
与前人研究对比分析, 发现本研究所鉴定15个稳定SNP位点中有8个位于前人定位的QTL区间, 4个SNP位点与前人所鉴定相关SNP物理距离小于2 Mb。如对于株高性状, 1号染色体稳定SNP位点PZE-101058322和PZE-101109358分别位于Zhou等[40]定位QTL区间和杨晓军等[41]定位区间。4号染色体SNP标记PZE-104088728和PZE-104102768分别位于Zhang等[42]定位QTL区间和Zhang等[42]定位QTL区间, 同时该位点与Hu等[43]定位SNP位点PZE-104103747仅相距1.15 Mb。5号染色体SNP标记PZE-105102182位于Tang等[44]定位区间, 同时该位点与Liu等[45]定位SNP位点chr5.s_156946316仅相距0.78 Mb。对于穗位高性状, 1号染色体SNP位点PZE-101024808与Dell’Acqua等[46]定位SYN12547位点相距1.90 Mb, 而另一个1号染色体SNP位点PZE-101105515同时位于Li等[28]、Tang等[44]和Park等[47]定位QTL区间和Pan等[48]所关联SNP位点1.92 Mb距离位置。5号染色体PZE-105098019同时位于杨晓军等[41]、Li等[28]以及Li等[49]定位的QTL区间。8号染色体PZE-108069615位点位于Park等[47]定位的QTL区间。由于目前对穗位系数性状研究较少, 仅发现8号染色体SNP位点PZE-108069615位于Wang等[19]定位的QTL区段。
综上, 本研究鉴定的15个稳定SNP位点中, 共有9个位点与前人定位研究结果相吻合, 表明本研究关联结果具有较高可信度, 所挖掘SNP可为分子标记辅助选择育种提供依据。同时发现一些SNP位点与前人多个独立研究定位结果相吻合, 暗示该位点附近可能存在可在不同环境稳定调控株高或穗位高性状的重要基因, 是后续克隆穗位高调控基因的重要目标区域。
6个稳定SNP位点为本研究所首次发现, 包括3个株高性状SNP位点, 其中在BLUP和20PG、21ZC环境中均检测到PZE-101256370, 且该位点PVE最高, 为主效SNP (表3)。另外3个SNP新位点分别为穗位系数显著关联PZE-103075978、PZE- 103083718和PZE-108039693, 其中PZE-103075978最高可解释5.58%表型变异, 属于主效SNP。这些株高和穗位系数相关新位点可同时在不同环境下, 具有较高可信度, 有助于进一步解析相关性状的遗传结构。
鉴定重要遗传位点优异等位变异有助于利用分子标记辅助选择将多个优异等位基因进行聚合, 从而培育优异种质。如李等[50]通过分子标记辅助选择与传统育种手段相结合, 实现了玉米抗南方锈病、抗茎腐病以及抗矮花叶病等多个性状的聚合育种。本研究对株高、穗位高和穗位系数性状的主效SNP位点进行分析, 发现该6个位点不同等位变异在4个环境间表型均具有显著差异, 优异等位变异可分别显著降低株高、穗位高及穗位系数, 说明以上主效SNP位点可用于分子标记辅助选择育种。此外, 本研究检测到一个可协同调控玉米株高、穗位高及穗位系数性状的SNP位点PZE-105102182, 该位点的优异等位变异可分别使株高、穗位高、穗位系数在4个环境下都得到显著降低, 因此该位点具有一因多效性, 可作为分子标记协同改良上述性状。
3.2 候选基因分析
目前已克隆到多个株高、穗位高相关性状调控基因, 主要包括激素相关基因, 如生长素(auxin)、赤霉素(gibberellin, GA)、油菜素内酯(brassinolide, BR)、独脚金内酯(strigolactone, SL)等, 同时微管发育、糖代谢、纤维素合成、根毛生长、转录因子调控等过程均可影响植株茎秆发育[33]。由此, 根据已克隆基因文献报道预测了稳定SNP位点置信区间的最可能候选基因。结果显示株高性状候选区间内存在有较多可能与茎秆发育相关的候选基因, 如PZE-101256370位点候选基因编码核质转运蛋白。Xu等[51]发现拟南芥Exportin1A (XPO1A)作为一种核质转运蛋白, 参与脱落酸(ABA)信号转导, 戚义东等[52]发现脱落酸可通过拮抗赤霉素降低水稻株高, 因此推测候选基因可能通过ABA途径参与株高性状调控。糖类在植物株高发育中扮演着重要角色, 为细胞分裂、伸长等生命活动提供了必要的物质基础, 如已克隆株高性状调控基因, 其突变体叶中因不能正常输出蔗糖, 导致植株表现矮小[53]。候选基因编码甘露糖苷酶, 其参与甘露糖苷键的水解过程, 甘露糖是一种单糖, 为多种多糖的组成成分, 由此推测该候选基因可能通过影响糖类物质合成进而影响株高性状。
穗位高性状关联位点PZE-105098019候选区间内基因编码泛素连接酶, 泛素-蛋白酶体途径在生长素、赤霉素、油菜素内酯等影响茎秆发育的激素信号转导中具有重要作用[54], 由此推测可能通过参与激素信号传导。PZE-101024808候选区间基因编码岩藻糖基转移酶蛋白, 冯玥[55]研究发现, 棉花岩藻糖基转移酶家族基因对于细胞伸长具有重要作用, 推测可能同样通过调控细胞伸长进而影响穗位高性状。
针对穗位系数性状, 候选基因中鉴定出较多转录因子编码基因(表3)。如PZE-103075978位点候选基因编码Dof锌指蛋白, Skirycz等[56]研究发现, 过表达拟南芥Dof转录因子基因, 会影响植物的细胞大小和数量, 造成植株矮化, 推测可能通过影响细胞大小和数量, 进而调控穗位系数性状。PZE-108069615是穗位高和穗位系数性状共定位的SNP位点, 该位点候选基因编码CPP转录因子, 而CPP转录因子参与花器官的发育, 在控制生殖组织发育和细胞分裂过程中起着极其重要的作用[57], 推测可能通过影响生殖组织发育来影响果穗的形成, 从而影响穗位高和穗位系数性状。此外, 本研究定位到一个一因多效位点PZE- 105102182, 该位点的优异等位变异可使株高、穗位高、穗位系数均得到显著降低, 因此挖掘该位点的候选基因对协同改良植物株型具有重要意义。该位点候选基因编码丝氨酸/苏氨酸磷酸酶, 是参与催化蛋白质去磷酸化的主要酶类之一, 可参与激素信号转导途径[58-59], 如已克隆玉米株高调控基因编码的丝氨酸/苏氨酸激酶参与油菜素内酯的信号传导, 其功能缺失突变体植株由于节间缩短而表现为矮小[60], 因此推测可能通过调控与油菜素内酯的信号传导调控玉米株型性状, 其调控功能仍有待进一步验证。
4 结论
本研究以854份自交系作为关联群体, 利用2795个SNP标记, 采用FarmCPU模型针对株高、穗位高、穗位系数性状进行全基因组关联分析, 共检测到81个显著关联SNP。在2个及以上环境中被稳定检测到的稳定SNP位点为15个, 其中6个SNP为本研究首次发现。在稳定SNP位点上下游各200 kb置信区间内, 根据基因功能注释及文献资料, 筛选出各位点的最可能候选基因, 这些候选基因主要参与激素合成与信号转导、糖类代谢、细胞分裂调控等途径。针对3个性状分别鉴定出2个主效SNP位点, 并鉴定出1个可同时调控株高、穗位高、穗位系数的一因多效位点。本研究可为分子标记辅助选择育种提供重要遗传位点, 为株高相关性状基因精细定位和克隆提供参考和依据。
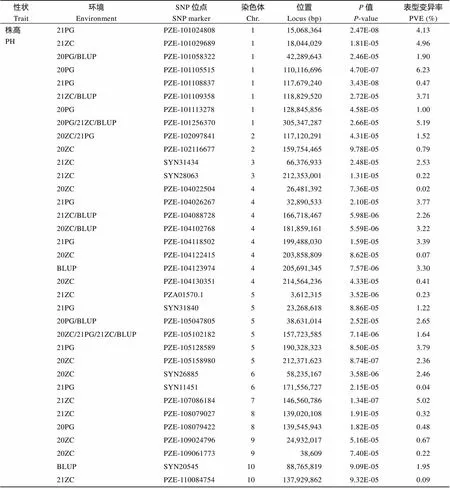
附表1 不同环境下株高、穗位高、穗位系数显著关联SNP位点汇总
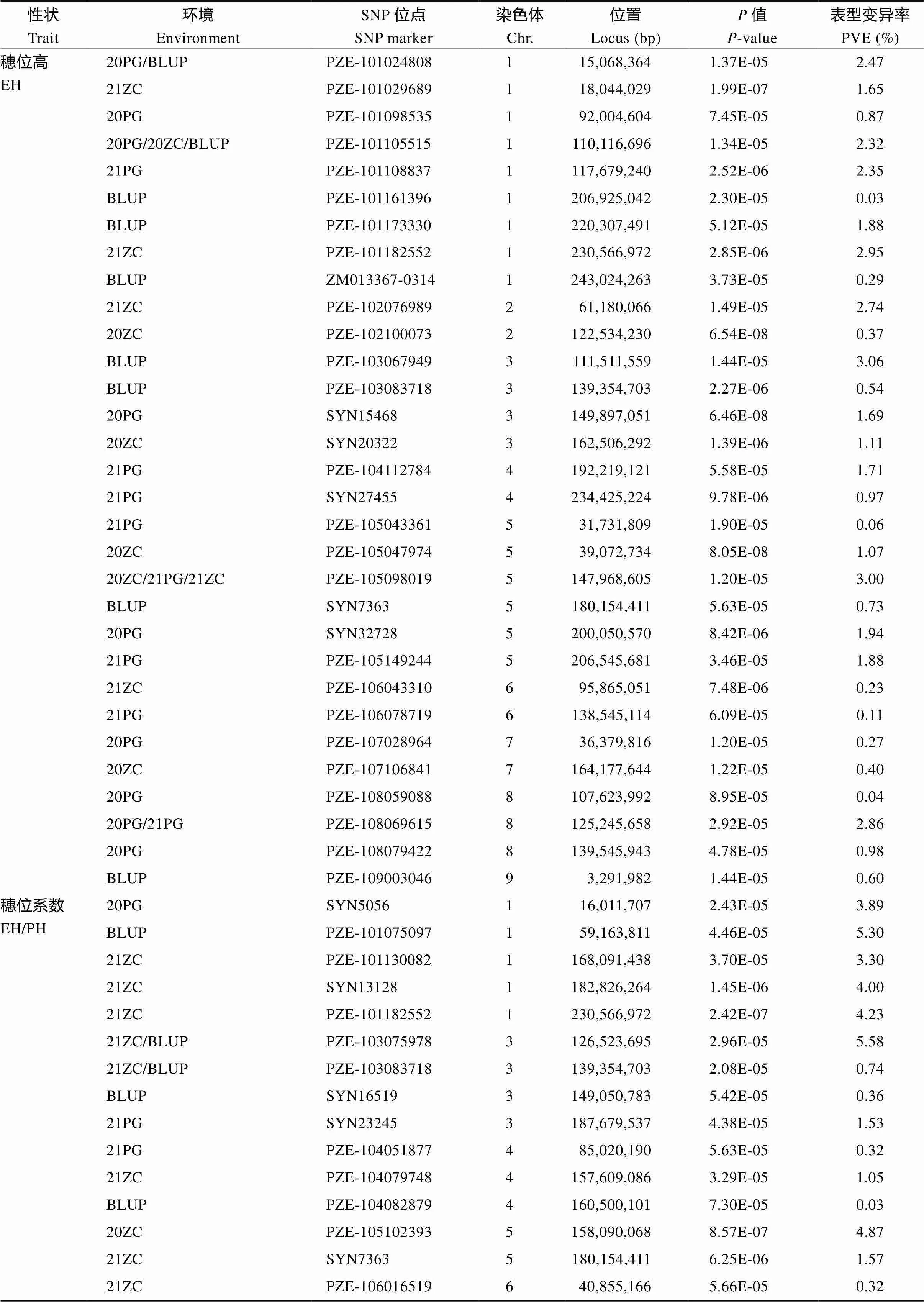
(续附表1)
PH、EH、EH/PH和BLUP分别表示株高、穗位高、穗位系数和最佳线性无偏预测值; PG、ZC分别表示平谷和诸城。
PH, EH, EH/PH, and BLUP represent plant height, ear height, the ratio of EH to PH and best linear unbiased prediction, respectively; PG and ZCrepresent Pinggu and Zhucheng, respectively.
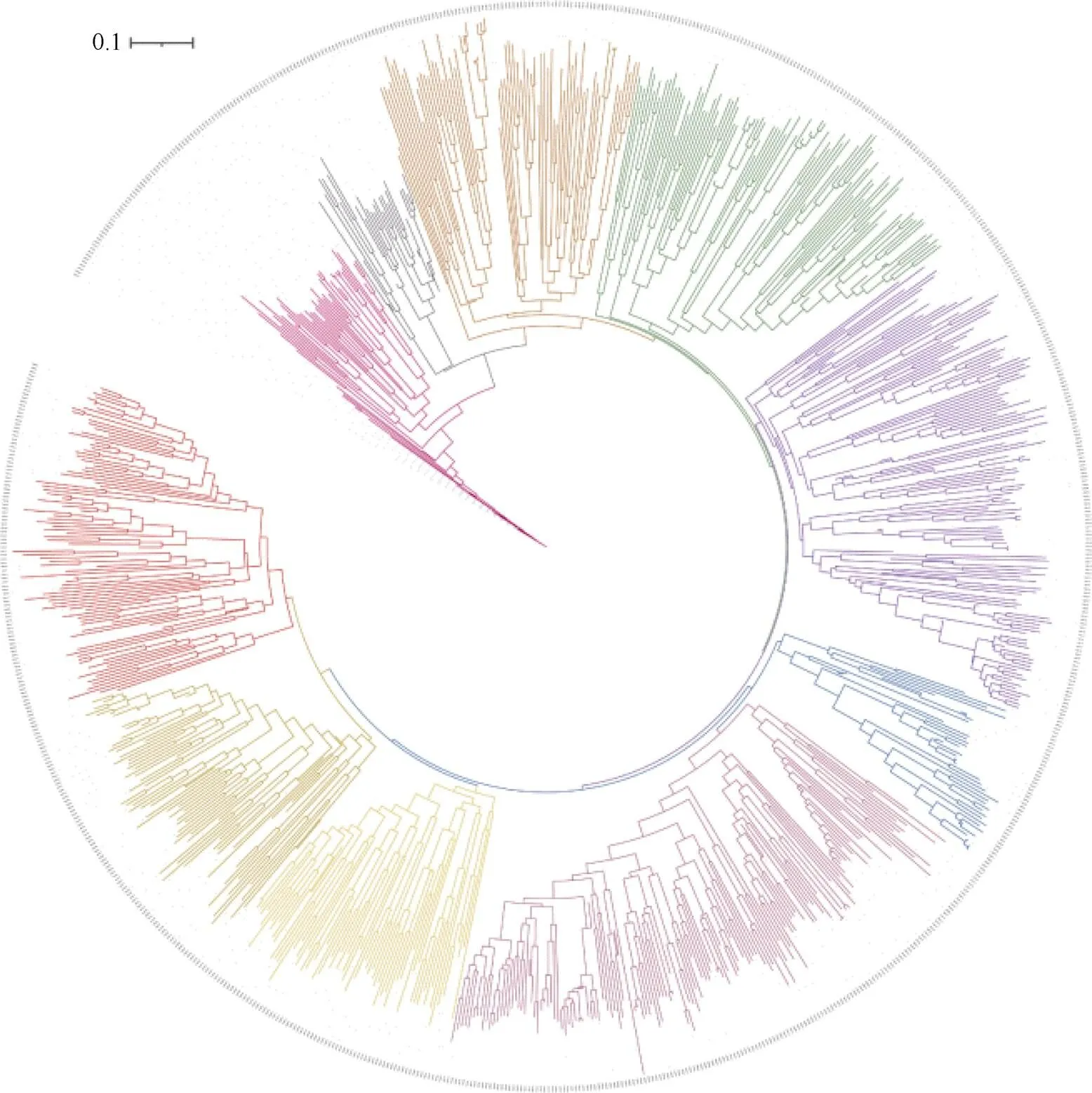
附图1 关联分析群体系统进化树
Fig. S1 Phylogenetic tree of the association analysis population
[1] Prasanna B M. Diversity in global maize germplasm: characterization and utilization., 2012, 37: 843–855.
[2] Tang J H, Teng W T, Yan J B, Ma X Q, Meng Y J, Dai J R, Li J S. Genetic dissection of plant height by molecular markers using a population of recombinant inbred lines in maize., 2007, 155: 117–124.
[3] 徐田军, 张勇, 赵久然, 王荣焕, 吕天放, 刘月娥, 蔡万涛, 刘宏伟, 陈传永, 王元东. 宜机收籽粒玉米品种冠层结构、光合及灌浆脱水特性. 作物学报, 2022, 48: 1526–1536.
Xu T J, Zhang Y, Zhao J R, Wang R H, Lyu T F, Liu Y E, Cai W T, Liu H W, Chen C Y, Wang Y D. Canopy structure, photosynthesis, grain filling, and dehydration characteristics of maize varieties suitable for grain mechanical harvesting.,2022, 48: 1526–1536 (in Chinese with English abstract).
[4] 崔爱民, 张久刚, 张虎, 单皓, 陈伟. 我国玉米生产现状及发展变革. 中国农业科技导报, 2020, 22(7): 10–19.
Cui A M, Zhang J G, Zhang H, Shan H, Chen W. Preliminary exploration on current situation and development of maize production in China., 2020, 22(7): 10–19 (in Chinese with English abstract).
[5] 宋振伟, 齐华, 张振平, 钱春荣, 郭金瑞, 邓艾兴, 张卫建. 春玉米中单909农艺性状和产量对密植的响应及其在东北不同区域的差异. 作物学报, 2012, 38: 2267–2277.
Song Z W, Qi H, Zhang Z P, Qian C R, Guo J R, Deng A X, Zhang W J. Effects of plant density on agronomic traits and yield in spring maize Zhongdan 909 and their regional differences in northeast China., 2012, 38: 2267–2277 (in Chinese with English abstract).
[6] Khush G S. Green revolution: the way forward., 2001, 2: 815–822.
[7] Donald C M. The breeding of crop ideotypes., 1968, 17: 385–403.
[8] Salas Fernandez M G, Becraft P W, Yin Y H, Lübberstedt T. From dwarves to giants? Plant height manipulation for biomass yield., 2009, 14: 454–461.
[9] 郑德波, 杨小红, 李建生, 严建兵, 张士龙, 贺正华, 黄益勤. 基于SNP标记的玉米株高及穗位高QTL定位. 作物学报, 2013, 39: 549–556.
Zheng D B, Yang X H, Li J S, Yan J B, Zhang S L, He Z H, Huang Y Q. QTL identification for plant height and ear height based on SNP mapping in maize (L.).,2013, 39: 549–556 (in Chinese with English abstract).
[10] 薛军, 王克如, 谢瑞芝, 勾玲, 张旺锋, 明博, 侯鹏, 李少昆. 玉米生长后期倒伏研究进展. 中国农业科学, 2018, 51: 1845–1854.
Xue J, Wang K R, Xie R Z, Gou L, Zhang W F, Ming B, Hou P, Li S K. Research progress of maize lodging during late stage.2018, 51: 1845–1854 (in Chinese with English abstract).
[11] 刘忠祥, 杨梅, 殷鹏程, 周玉乾, 何海军, 邱法展. 玉米株高主效QTL精细定位及遗传效应分析. 作物学报, 2018, 44: 1357–1366.
Liu Z X, Yang M, Yin P C, Zhou Y Q, He H J, Qiu F Z. Fine mapping and genetic effect analysis of a major QTLassociated with plant height in maize (L.)., 2018, 44: 1357–1366 (in Chinese with English abstract).
[12] 刘磊, 詹为民, 丁武思, 刘通, 崔连花, 姜良良, 张艳培, 杨建平. 玉米矮化突变体的遗传分析与分子鉴定. 作物学报, 2022, 48: 886–895.
Liu L, Zhan W M, Ding W S, Liu T, Cui L H, Jiang L L, Zhang Y P, Yang J P. Genetic analysis and molecular characterization of dwarf mutantin maize., 2022, 48: 886–895 (in Chinese with English abstract).
[13] 于芮苏, 田小康, 刘斌斌, 段迎新, 李婷, 张秀英, 张兴华, 郝引川, 李勤, 薛吉全, 徐淑兔. 玉米抗倒伏相关性状QTL的关联和连锁分析. 作物学报, 2022, 48: 138–150.
Yu R S, Tian X K, Liu B B, Duan Y X, Li T, Zhang X Y, Zhang X H, Hao Y C, Li Q, Xue J Q, Xu S T. Dissecting the genetic architecture of lodging related traits by genome-wide association study and linkage analysis in maize., 2022, 48: 138–150 (in Chinese with English abstract).
[14] Beavis W D, Grant D, Albertsen M, Fincher R. Quantitative trait loci for plant height in four maize populations and their associations with qualitative genetic loci., 1991, 83: 141–145.
[15] Yan J B, Tang H, Huang Y Q, Shi Y G, Li J S, Zheng Y L. Dynamic analysis of QTL for plant height at different developmental stages in maize (L.)., 2003, 48: 2601–2607.
[16] Weng J F, Xie C X, Hao Z F, Wang J J, Liu C L, Li M S, Zhang D G, Bai L, Zhang S H, Li X H. Genome-wide association study identifies candidate genes that affect plant height in Chinese elite maize (L.) inbred lines., 2011, 6: e29229.
[17] Bai W, Zhang H, Zhang Z, Teng F, Wang L, Tao Y, Zheng Y. The evidence for non-additive effect as the main genetic component of plant height and ear height in maize using introgression line populations., 2009, 129: 376–384.
[18] Vanous A, Gardner C, Blanco M, Blanco M, Schwarze A M, Lipka A E, Garcia S F, Bohn M, Edward J, Lübberstedt T. Association mapping of flowering and height traits in germplasm enhancement of maize doubled haploid (GEM-DH) lines., 2018, 11: 170083.
[19] Wang B B, Liu H, Liu Z P, Dong X M, Guo J J, Li W, Chen J, Gao C, Zhu Y B, Zheng X M, Chen Z L, Chen J, Song W B, Hauck A, Lai J S. Identification of minor effect QTLs for plant architecture related traits using super high density genotyping and large recombinant inbred population in maize ()., 2018, 18: 17.
[20] 刘坤, 张雪海, 孙高阳, 闫鹏帅, 郭海平, 陈思远, 薛亚东, 郭战勇, 谢惠玲, 汤继华, 李卫华. 玉米株型相关性状的全基因组关联分析. 中国农业科学, 2018, 51: 821–834.
Liu K, Zhang X H, Sun G Y, Yan P S, Guo H P, Chen S Y, Xue Y D, Guo Z Y, Xie H L, Tang J H, Li W H. Genome-wide association studies of plant type traits in maize., 2018, 51: 821–834 (in Chinese with English abstract).
[21] Knapp S J, Stroup W W, Ross W M. Exact confidence intervals for heritability on a progeny mean basis., 1985, 25: 192–194.
[22] Chen D H, Ronald P C. A rapid DNA minipreparation method suitable for AFLP and other PCR application., 1999, 17: 53–57.
[23] Tian H L, Wang F G, Zhao J R, Yi H M, Wang L, Wang R. Yang Y, Song W. Development of maize SNP3072, a high-throughput compatible SNP array, for DNA fingerprinting identification of Chinese maize varieties., 2015, 35: 136.
[24] Pritchard J K, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data., 2000, 155: 945–959.
[25] Liu X L, Huang M, Fan B, Buckler E S, Zhang Z W. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies., 2016, 12: e1005767.
[26] Wang M, Yan J B, Zhao J R, Song W, Zhang X B, Xiao Y N, Zheng Y L. Genome-wide association study (GWAS) of resistance to head smut in maize., 2012, 196: 125–131.
[27] 贺建波, 刘方东, 邢光南, 王吴彬, 赵团结, 管荣展, 盖钧镒. 限制性两阶段多位点全基因组关联分析方法的特点与计算程序. 作物学报, 2018, 44: 1274–1289.
He J B, Liu F D, Xing G N, Wang W B, Zhao T J, Guan R Z, Gai J Y. Characterization and analytical programs of the restricted two-stage multi-locus genome-wide association analysis., 2018, 44: 1274–1289 (in Chinese with English abstract).
[28] Li X P, Zhou Z J, Ding J Q, Wu Y B, Zhou B, Wang R X, Ma J L, Wang S W, Zhang X C, Xia Z L, Chen J F, Wu J Y. Combined linkage and association mapping reveals QTL and candidate genes for plant and ear height in maize., 2016, 7: 833.
[29] 李博, 张焕欣, 杨小艳, 吕颖颖, 江培顺, 郝转芳, 吕香玲, 王宏伟, 翁建峰. 玉米穗位高全基因组关联分析及其候选基因预测. 作物杂志, 2013, (2): 27–32.
Li B, Zhang H X, Yang X Y, Lyu Y Y, Jiang P S, Hao Z F, Lyu X L, Wang H W, Weng J F. Genome-wide association study and candidate gene prediction of ear height in maize (L.)., 2013, (2): 27–32 (in Chinese with English abstract).
[30] 张焕欣, 翁建峰, 张晓聪, 刘昌林, 雍洪军, 郝转芳, 李新海. 玉米穗行数全基因组关联分析. 作物学报, 2014, 40: 1–6.
Zhang H X, Weng J F, Zhang X C, Liu C L, Yong H J, Hao Z F, Li X H. Genome-wide association analysis of kernel row number in maize., 2014, 40: 1–6 (in Chinese with English abstract).
[31] Pandis N. Linear regression., 2016, 149: 431–434.
[32] 袁亮, 孟鑫, 汪亚龙, 廖长见, 李高科, 吕桂华, 宋军, 邱正高, 林海建. 镉胁迫下甜、糯玉米开花期性状的全基因组关联分析. 植物遗传资源学报, 2021, 22: 438–447.
Yuan L, Meng X, Wang Y L, Liao C J, Li G K, Lyu G H, Song J, Qiu Z G, Lin H J. Genome wide association analysis of flowering traits in sweet and waxy maize under cadmium stress., 2021, 22: 438–447 (in Chinese with English abstract).
[33] 马雅杰, 高悦欣, 李依萍, 龙艳, 董振营, 万向元. 玉米株高和穗位高的遗传基础与分子机制. 中国生物工程杂志, 2021, 41(12): 61–73.
Ma Y J, Gao Y X, Li Y P, Long Y, Dong Z Y, Wan X Y. Progress on genetic analysis and molecular dissection on maize plant height and ear height., 2021, 41(12): 61–73 (in Chinese with English abstract).
[34] Ertiro B T, Labuschagne M, Olsen M, Das B, Prasanna B M, Gowda M. Genetic dissection of nitrogen use efficiency in tropical maize through genome-wide association and genomic prediction., 2020, 11: 474.
[35] Zhu C, Gore M, Buckler E S, Yu J M. Status and prospects of association mapping in plants., 2008, 1: 5–20.
[36] An Y X, Chen L, Li Y X, Li C H, Shi Y S, Zhang D F, Li Y, Wang T Y. Genome-wide association studies and whole-genome prediction reveal the genetic architecture of KRN in maize., 2020, 20: 490.
[37] Zhang Y, Wan J Y, He L, Lan H, Li L J. Genome-wide association analysis of plant height using the maize F1population.(Basel), 2019, 8: 432.
[38] 渠建洲, 冯文豪, 张兴华, 徐淑兔, 薛吉全. 基于全基因组关联分析解析玉米籽粒大小的遗传结构. 作物学报, 2022, 48: 304–319.
Qu J Z, Feng W H, Zhang X H, Xu S T, Xue J Q. Dissecting the genetic architecture of maize kernel size based on genome-wide association study., 2022, 48: 304–319 (in Chinese with English abstract).
[39] Zhao Y, Wang H S, Bo C, Dai W, Zhang X G, Cai R H, Gu L J, Ma Q, Jiang H Y, Zhu J, Cheng B J. Genome-wide association study of maize plant architecture using F1populations., 2019, 99: 1–15.
[40] Zhou Z Q, Zhang C S, Lu X H, Wang L W, Hao Z F, Li M S, Zhang D G, Yong H J, Zhu H Y, Weng J F, Li X H. Dissecting the genetic basis underlying combining ability of plant height related traits in maize., 2018, 9: 1117.
[41] 杨晓军, 路明, 张世煌, 周芳, 曲延英, 谢传晓. 玉米株高和穗位高的QTL定位. 遗传, 2008, 30: 1477–1486.
Yang X J, Lu M, Zhang S H, Zhou F, Qu Y Y, Xie C X. QTL mapping of plant height and ear position in maize (L.).(Beijing), 2008, 30: 1477–1486 (in Chinese with English abstract).
[42] Zhang Z M, Zhao M J, Ding H P, Rong T Z, Pan G T. Quantitative trait loci analysis of plant height and ear height in maize (L.)., 2006, 42: 306–310.
[43] Hu S L, Wang C L, Sanchez D L, Lipka A E, Liu P, Yin Y H, Blanco M, Lübberstedt T. Gibberellins promote brassinosteroids action and both increase heterosis for plant height in maize (L.)., 2017, 8: 1039.
[44] Tang Z X, Yang Z F, Hu Z Q, Zhang D, Lu X, Jia B, Deng D X, Xu C W. Cytonuclear epistatic quantitative trait locus mapping for plant height and ear height in maize., 2013, 31: 1–14.
[45] Liu H J, Wang X Q, Xiao Y J, Luo J Y, Qiao F, Yang W Y, Zhang R Y, Meng Y J, Sun J M, Yan S J, Peng Y, Niu L Y, Jian L M, Song W, Yan J L, Li C H, Zhao Y X, Liu Y, Warburton M L, Zhao J R, Yan J B. CUBIC: an atlas of genetic architecture promises directed maize improvement., 2020, 21: 20.
[46] Dell’Acqua M, Gatti D M, Pea G, Cattonaro F, Coppens F, Magris G, Hlaing A L, Aung H H, Nelissen H, Baute J, Frascaroli E, Churchill G A, Inzé D, Morgante M, Pè M E. Genetic properties of the MAGIC maize population: a new platform for high definition QTL mapping in., 2015, 16: 167.
[47] Park K J, Sa K J, Kim B W, Koh H J, Lee J K. Genetic mapping and QTL analysis for yield and agronomic traits with an F2:3population derived from a waxy corn × sweet corn cross., 2014, 36: 179–189.
[48] Pan Q C, Xu Y C, Li K, Peng Y, Zhan W, Li W Q, Li L, Yan J B. The genetic basis of plant architecture in 10 maize recombinant inbred line populations., 2017, 175: 858–873.
[49] Li Y L, Dong Y B, Niu S Z, Cui D Q. The genetic relationship among plant-height traits found using multiple-trait QTL mapping of a dent corn and popcorn cross., 2007, 50: 357–364.
[50] 李卫华. 玉米多种抗病基因的分子聚合育种. 中国科学院遗传与发育生物学研究所博士学位论文, 北京, 2008.
Li W H. Pyramiding Breeding of Resistance Genes to Maize Diseases with Marker-assisted Selection. PhD Dissertation of Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing, China, 2008 (in Chinese with English abstract).
[51] Xu X Z, Wan W, Jiang G B, Xi Y, Huang H J, Cai J J, Chang Y N, Duan C G, Mangrauthia S K, Peng X X, Zhu J K, Zhu G H. Nucleocytoplasmic trafficking of theWD40 repeat protein XIW1 regulates ABI5 stability and abscisic acid responses., 2019, 12: 1598–1611.
[52] 戚义东, 秦华, 高雅迪, 王芳芳, 黄荣峰, 权瑞党. 脱落酸拮抗赤霉素抑制水稻地上部生长的研究. 生物技术进展, 2019, 9: 483–489.
Qi Y D, Qin H, Gao Y D, Wang F F, Huang R F, Quan R D. Study on antagonizing regulation of shoot growth by abscisic acid and gibberellic acid in rice., 2019, 9: 483–489 (in Chinese with English abstract).
[53] Russin W A, Evert R F, Vanderveer P J, Sharkey T D, Briggs S P. Modification of a specific class of plasmodesmata and loss of sucrose export ability in themaize mutant., 1996, 8: 645–658.
[54] 覃碧. 泛素蛋白酶体途径及其对植物激素信号转导的调控. 热带农业科学, 2013, 33: 39–45.
Qin B. Ubiquitin-proteasome pathway and its regulation of plant hormone signaling., 2013, 33: 39–45 (in Chinese with English abstract).
[55] 冯玥. 棉花岩藻糖基转移酶家族基因()的发掘和功能初步分析. 南京农业大学博士学位论文, 江苏南京, 2016.
Feng Y. Genome-wide Identification of Fucosyltransferase () Gene Family and Functional Analysis ofin Cotton. PhD Dissertation of Nanjing Agricultural University, Nanjing, Jiangsu, China, 2016 (in Chinese with English abstract).
[56] Skirycz A, Radziejwoski A, Busch W, Hannah M A, Czeszejko J, Kwasniewski M, Zanor M I, Lohmann J U, Veylder L D, Witt I, Roeber B M. The DOF transcription factor OBP1 is involved in cell cycle regulation in., 2008, 56: 779–792.
[57] 王凯. 拟南芥和水稻CPP转录因子家族的生物信息学分析. 生物技术通报, 2010, (2): 76–84. Wang K. Bioinformatic analysis of the CPP transcription factors family inand rice., 2010, (2): 76–84 (in Chinese with English abstract).
[58] 何亮, 李富华, 沙莉娜, 付凤玲, 李晚忱. 玉米2C型丝氨酸/苏氨酸蛋白磷酸酶(PP2C)活性与耐旱性的关系. 作物学报, 2008, 34: 899–903.
He L, Li F H, Sha L N, Fu F L, Li W C. Activity of serine/threonine protein phosphatase type-2C (PP2C) and its relationships to drought tolerance in maize., 2008, 34: 899–903 (in Chinese with English abstract).
[59] 卫卓赟, 黎家. 受体激酶介导的油菜素内酯信号转导途径. 生命科学, 2011, 23: 1106–1113.
Wei Z Y, Li J. Receptor kinases mediated brassinosteroid signal transduction in plants., 2011, 23: 1106–1113 (in Chinese with English abstract).
[60] Kir G, Ye H X, Nelissen H, Neelakandan A K, Kusnandar A S, Luo A, Inzé D, Sylvester A W, Yin Y H, Becraft P W. RNA interference knockdown ofin maize reveals novel functions for brassinosteroid signaling in controlling plant architecture., 2015, 169: 826–839.
Genome-wide association analysis of plant height and ear height related traits in maize
MA Ya-Jie1,**, BAO Jian-Xi1,**, GAO Yue-Xin1, LI Ya-Nan1, QIN Wen-Xuan1, WANG Yan-Bo1, LONG Yan1, LI Jin-Ping2, DONG Zhen-Ying1,2,*, and WAN Xiang-Yuan1,2,*
1Zhongzhi International Institute of Agricultural Biosciences, Shunde Graduate School, School of Chemistry and Biological Engineering, Research Center of Biology and Agriculture, University of Science and Technology Beijing (USTB), Beijing 100083, China;2Beijing Engineering Laboratory of Main Crop Bio-Tech Breeding, Beijing International Science and Technology Cooperation Base of Bio-Tech Breeding, Beijing Solidwill Sci-Tech Co. Ltd., Beijing 100192, China
Suitable plant height (PH) and ear height (EH) can improve the efficiency of nutrient utilization and lodging resistance, which is of great significance for stable and high yield in maize. In this study, an association panel including 854 maize inbred lines used to analyze the PH, EH, and the ratio of EH to PH (EH/PH) in four environments, and genome-wide association study (GWAS) was then conducted using 2795 single nucleotide polymorphism (SNP) markers distributed uniformly throughout maize genome. A total of 81 SNP loci (< 0.0001) were identified by using FarmCPU model, among which 35 SNPs were significantly associated with PH, with phenotypic variation explained (PVE) ranging from 0.020% to 6.225%; 31 SNPs were significantly associated with ear height, and PVE was from 0.026% to 3.060%; 24 SNPs were significantly associated with EH/PH, and the PVE ranged from 0.032% to 6.636%. 15 stable SNPs that were repeatedly detected in multiple environments for specific trait were further identified, among which six loci were reported for the first time in this study, and the remaining nine loci located in the previously identified quantitative trait loci (QTLs) or/and no more than 2 Mb with the known SNPs related with PH and EH traits. A total of 83 genes were annotated in the confidence intervals of the 15 stable SNPs, and the most likely candidate genes were further predicted according to the gene functional annotations and comparison with previous reports. The candidate genes were mainly involved in hormone synthesis and signal transduction, carbohydrate metabolism, cell division regulation and so on. Finally, six major SNP loci and one locus that affected PH, EH, and EH/PH simultaneously were identified. This study can provide genetic loci for molecular marker-assisted selection in maize breeding and provide valuable information for fine mapping and cloning of PH and EH related genes.
maize; plant height; ear height; genome-wide association analysis; candidate gene
10.3724/SP.J.1006.2023.23023
本研究由国家重点研发计划项目“农业生物种质资源挖掘与创新利用”重点专项(2021YFD1200700)资助。
This study was supported by the National Key Research and Development Program of China (2021YFD1200700).
通信作者(Corresponding authors):董振营, E-mail: zydong@ustb.edu.cn; 万向元, E-mail: wanxiangyuan@ustb.edu.cn
同等贡献(Contributed equally to this work)
马雅杰, E-mail: 13292097686@163.com; 鲍建喜, E-mail: bjx1232003@126.com
2022-03-03;
2022-05-05;
2022-05-19.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20220518.1923.002.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
猜你喜欢
作物学报(2022年8期)2022-05-29
作物学报(2022年6期)2022-04-08
作物杂志(2022年6期)2022-02-03
国际医学放射学杂志(2021年5期)2021-10-22
湖南农业大学学报(自然科学版)(2020年4期)2020-08-28
农村百事通(2019年17期)2019-10-08
山西农业大学学报(自然科学版)(2018年12期)2018-12-04
麦类作物学报(2018年4期)2018-05-11
第一财经(2017年36期)2017-09-25
现代农业科技(2017年1期)2017-03-06
