
基于分子体积法的CHONF高能致爆材料晶体密度的预测
2023-01-16 06:13:34文琳元何晓凯师进文刘英哲王伯周
火炸药学报 2022年6期
文琳元,何晓凯,师进文,刘英哲,王伯周
(1.西安交通大学 能源与动力工程学院,陕西 西安 710049;2.西安近代化学研究所,陕西 西安 710065)
引 言
作为炸药、发射药、推进剂和火工品中高能量组分的含能材料,其能量密度水平一定程度上决定了武器发射、推进及毁伤能力,乃至国家整体军事装备的战略威慑和打击能力。追求更高能量密度,成为了含能材料领域科学家为之长期奋斗的目标。为此,研究者们提出了引入高能致爆基团构筑新型高能量密度含能材料的策略[1]。同时,由于新材料的合成通常需要大量的时间与金钱,因此准确预测含能材料性能对于开发具有优良性能的新型含能材料至关重要。随着计算科学与理论化学的快速发展,新型含能材料的理论设计技术日渐成熟,具体为通过量化计算预估新化合物的生成焓、密度、爆轰速度、爆轰压力等参数,辅以性能指标要求筛选到最终的目标化合物。其中,因为K-J方程中爆轰速度与密度正相关且爆轰压力与密度的平方成正比,所以密度被认为是影响含能材料爆轰性能的关键参数,如何准确预测新型含能材料晶体密度也就成为新型含能材料研发中一大关键问题。
当前含能材料晶体密度预测方法包含物理模型与统计模型两种。其中物理模型可细分为基于分子体积[2-4]以及基于晶体体积[5-9]两类,而基于统计模型中较为主流的方法则是机器学习[10]。目前基于分子体积方法用的较多的是分子表面静电势法,该方法是用分子摩尔质量(M)除以使用Monte-Carlo方法计算得到的分子0.001a.u.电子密度等值面包裹内体积V0.001的值来表示晶体密度[11-12],该方法未考虑分子间作用和晶体堆积,计算误差与速度适中。Politzer 在分子表面静电势法的基础上,针对CHON类材料提出了分子表面静电势参数修正的方法[13-14],其预测密度与实验值误差小于0.05g/cm3,被广泛运用于含能材料晶体密度预测。
基于晶体体积方法则是通过晶体结构预测,以获得含能材料准确的晶体结构,从而得到该材料的晶体密度。该方法需要准确描述分子间相互作用,存在计算速度较慢的不足,是当前含能材料晶体密度研究的一大挑战。而机器学习模型主要是通过各种回归算法构建材料结构与密度之间的关系,其中大多数是黑箱模型,缺乏解释性。近年来,不少研究者基于该方法在CHON类含能材料晶体密度预测上做了许多优秀工作,其中张朝阳研究员针对超过2000个硝基化合物构建了均方根误差(RMSE)=0.049g/cm3的图神经网络密度预测模型[15],T. Yong-Jin Han针对10251个含有至少一根氮氧键的CHON类化合物开发了RMSE=0.062g/cm3消息传递神经网络模型[16],张庆华研究员对超过1000个CHON类含能材料利用核岭回归的方法构建了MAE=0.042g/cm3的密度预测模型[17]。
从上述研究可以发现,其样本量均大于103,这是因为机器学习模型的计算精度十分依赖数据样本,样本量过少会使得模型过拟合。这其中,符号回归作为一种回归分析方法,在挖掘变量间内在关系以及帮助理解物理机制并发现隐藏的数学表达式方面取得一定的成果[18-19]。
由于当前晶体密度预测方法大多针对CHON类含能材料,没有考虑氟代偕二硝基(—CF(NO2)2)、二氟氨基(—C(NF2))以及氟代偕硝基(—CF(NO2))等含氟高能致爆基团对密度预测的影响,难以保证上述方法在预测含氟高能致爆材料晶体密度的精度。因此,有必要建立一个适用于含氟高能致爆材料的晶体密度预估方法。其中,基于晶体体积方法存在计算速度较慢、缺乏准确描述分子间相互作用的不足,因此本研究仅采用基于分子体积与基于机器学习的方法。先在Politzer基于CHON类含能材料密度预测公式基础上,利用多元线性拟合方法得到适合含氟高能致爆材料体系的晶体密度预测公式。考虑到含有上述含氟高能致爆基团材料数量较少的情况,本研究将机器学习模型中的符号回归算法与基于分子体积方法相结合,从多种静电势参数中确定出更适合于该研究体系的变量表现形式,构造出了适用于含氟高能致爆材料体系的R2=0.77、RMSE=0.045g/cm3的晶体密度预测公式。所得公式能提高含氟高能致爆材料晶体密度预测精度,以期为含氟高能致爆材料的高效准确设计奠定基础。
1 流程及原理
从剑桥晶体结构数据库(CCDC)中,按照实验密度大于1.6g/cm3、晶体结构为单晶、分子中存在含氟高能致爆基团等标准,筛选出56个化合物。再利用Gaussian16[20]软件的B3PW91/6-31g(d,p)方法,对上述化合物的分子进行几何结构优化并得到能量最优的构型,经频率分析确定无虚频。
根据分子表面静电势理论,利用Multiwfn[21]计算得到各参数,表1列出几个典型静电势参数。基于多元线性拟合方法,对Politzer所提的密度拟合公式进行修正,并在静电势参数基础上采用符号回归方法得到最优的公式形式。
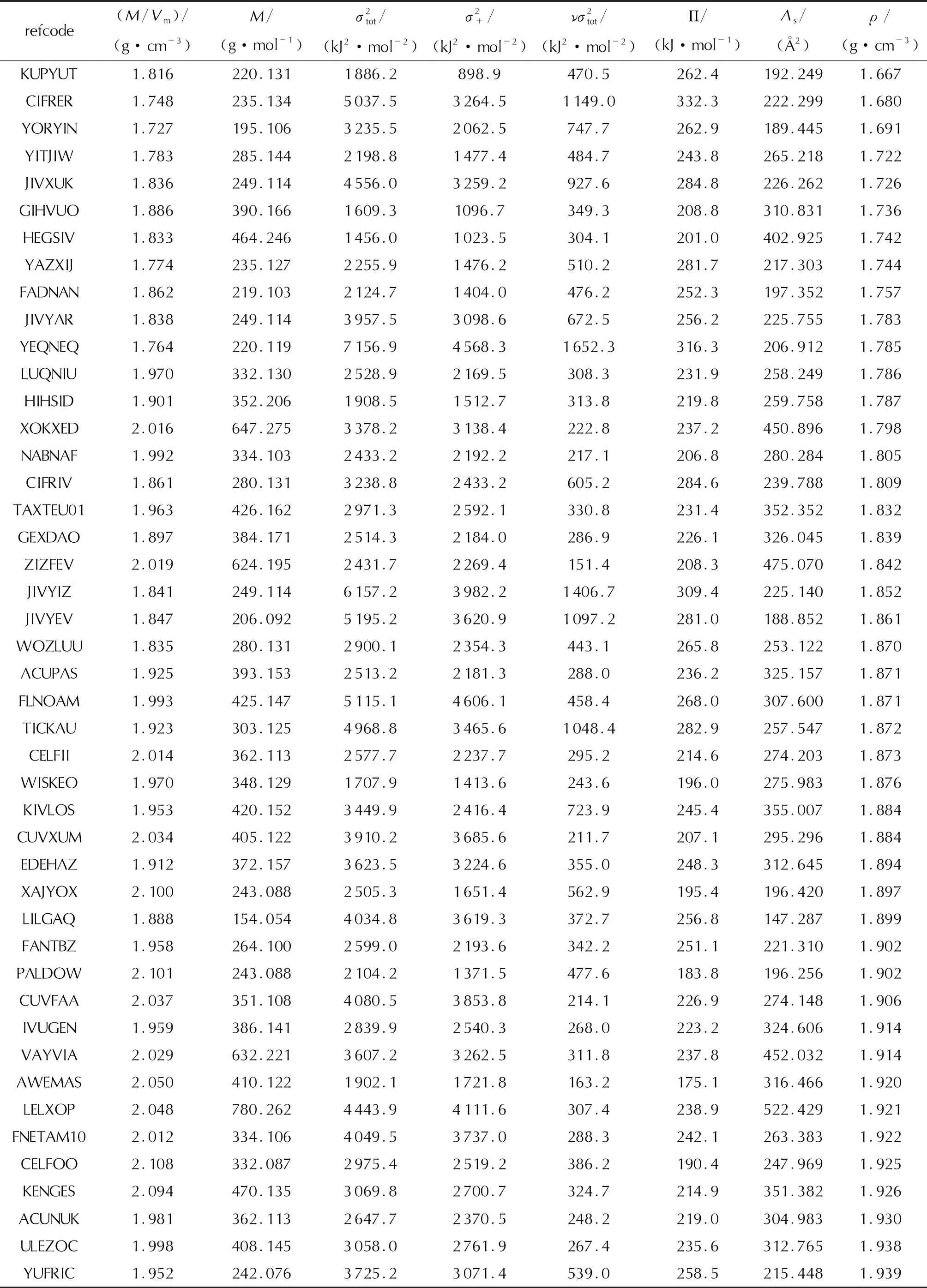
表1 56个含氟高能致爆材料的部分静电势参数及实验晶体密度Table 1 Electrostatic potential parameters and experimental crystal densities for 56 fluorine-containing high energetic explosophoric materials
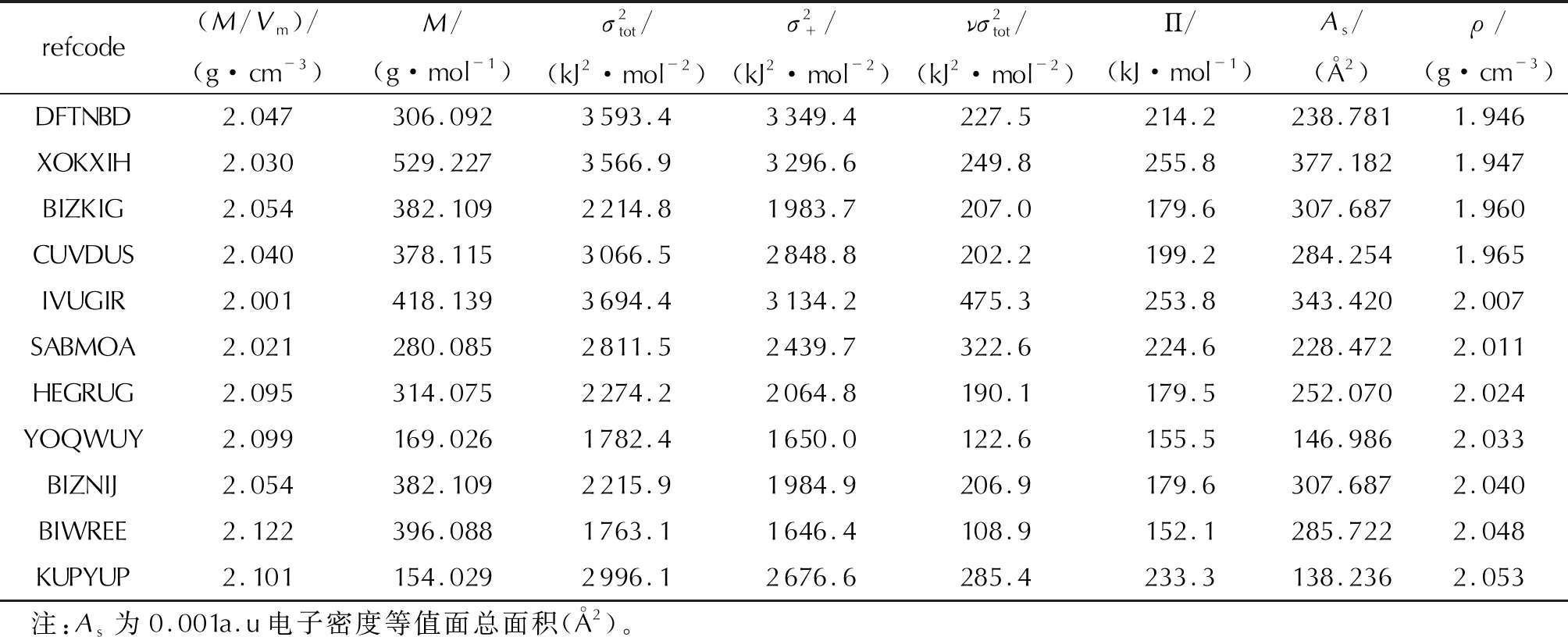
Continued
其中,选取了Rice在2007年所提的公式(1)[12]、Politzer在2009年所提的两个公式(2)和(3)[13]作为对比,并计算了各公式在预测含氟高能致爆材料时的均方根误差(RMSE)。
(1)
(2)
(3)
2 结果与讨论
根据公式(1),其实验晶体密度与预测密度关系如图1所示。
从图1可以清楚地发现,其预测密度几乎都大于对应的实验晶体密度,这首先说明氟元素加入改变了其分子间相互作用强弱,使得0.001a.u.电子密度等值面包裹的体积Vm小于等效的晶体体积;同时,按照Goh的标准[22],公式(1)计算中仅有17.9%的化合物预测密度可视为准确(误差小于 0.03g/cm3),25.0%的化合物预测密度可视为有用(误差小于0.05g/cm3),而公式(1)预测密度的均方根偏差高达0.106g/cm3,不足以达到晶体密度预测的要求。
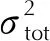
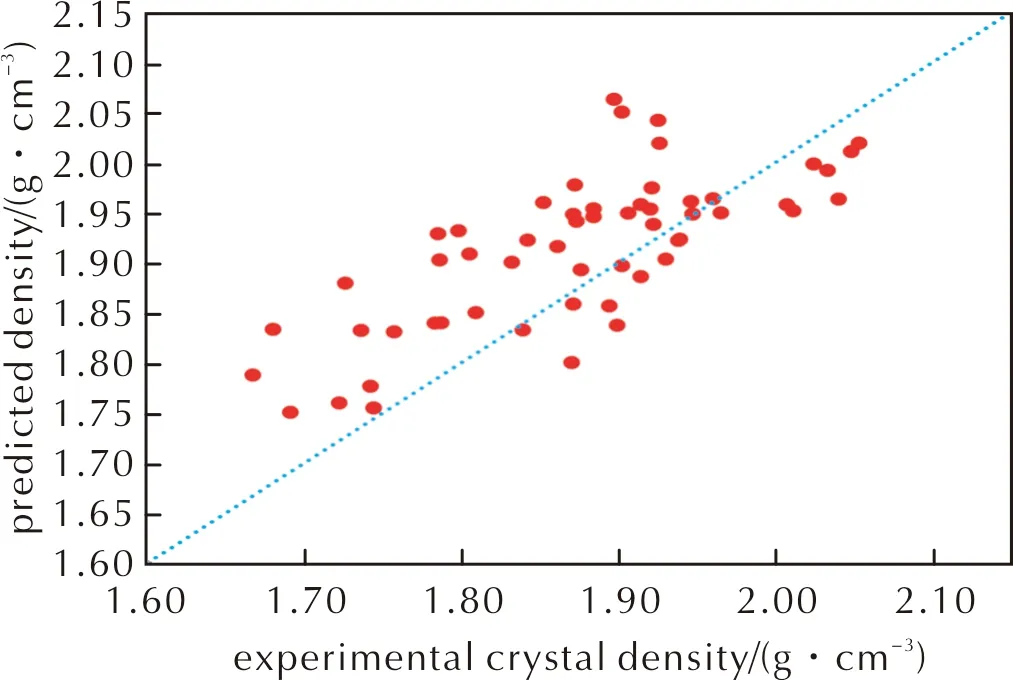
图公式下预测密度与实验晶体密度关系Fig.2 The relationship between predicted density and experimental crystal density under formula
从图1和图2可以看出,公式(2)得到预测密度比公式(1)预测密度小,这主要在于公式(2)中主要影响因素(M/Vm)前面的系数α小于公式(1)中的系数α=1。该公式使得预测结果更偏向于实际晶体密度,但从图2看,其仍呈现出高估了晶体密度的现象。相较没考虑分子间相互作用的公式(1),公式(2)计算得到的晶体密度与实际晶体密度的偏差减小,均方根偏差从0.106g/cm3降到0.075g/cm3,在56种含氟高能致爆材料中,预测晶体密度可视为准确的比例提高到26.8%,而视为有用的比例提高到44.6%,几乎是成倍地增长。这说明,加入静电势修正项,是符合物理本质且对于含氟晶体密度预测有用的。但是,其中仍有21.4%的结果偏差是大于0.100g/cm3,说明这一套仅考虑C、H、O、N元素的静电势修正拟合参数,不足以满足现有的需求。
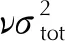
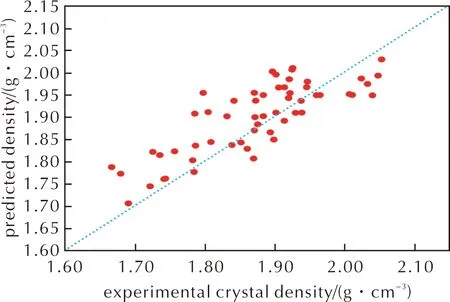
图3 Politer-Π公式下预测密度与实验晶体密度关系Fig.3 The relationship between predicted density and experimental crystal density under Politer-Π formula
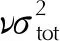
由于上述使用Politer所提的分子表面静电势修正公式在预测含氟高能致爆材料晶体密度时误差仍较大,因此为了更为准确地描述氟元素加入后分子间相互作用对预测公式的影响。综合考虑到(M/Vm)在公式(1)、(2)、(3)中关键作用,在公式(2)、(3)的基础采用多元线性拟合方法修正密度预测公式,得到如下式(4)、(5):
(4)
(5)
发现修正公式(4)和(5)的决定系数与均方根误差都为0.65和0.055g/cm3;而公式(4)在密度预测误差小于0.05g/cm3和小于0.03g/cm3上的表现优于公式(5),因此在后续仅讨论公式(4)的效果。其预测密度与实际晶体密度如图4所示,相较于前面提及的3种方法,改进后预测结果的相对误差都在5%以内,而且均方根偏差也降至0.055g/cm3。预测晶体密度可视为准确的比例为33.9%,而视为有用的比例提高到60.7%。不仅如此,预测晶体密度的偏差大于0.100g/cm3的比例也降到1.8%。
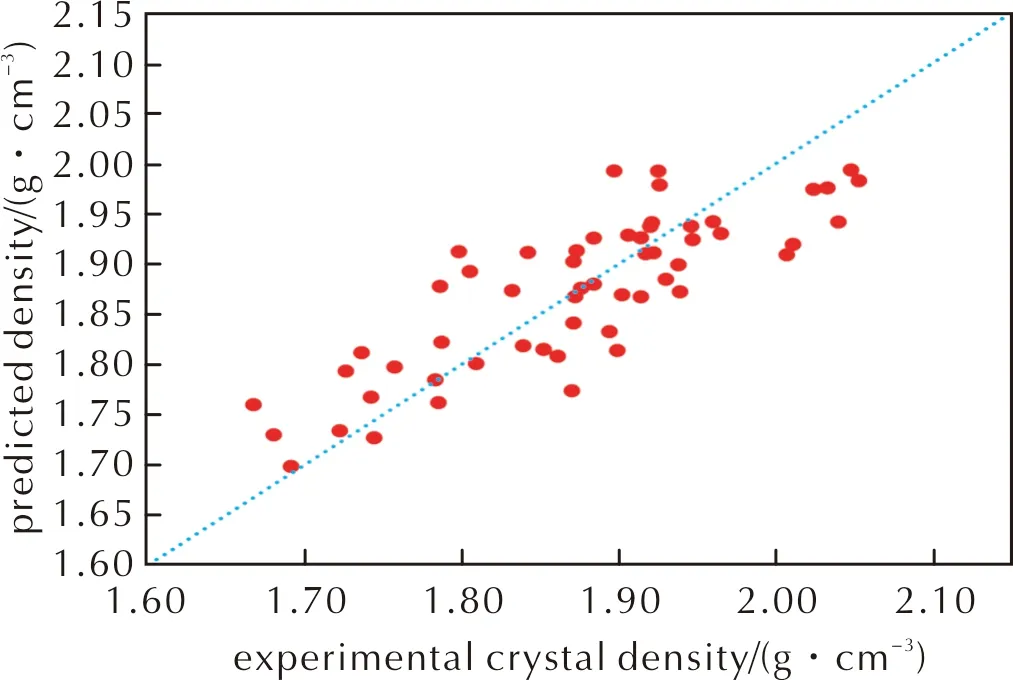
图4 多元线性拟合改进后密度预测公式中预测密度与实验晶体密度关系Fig.4 The relationship between predicted density and experimental crystal density in the improved density prediction formula by multiple linear regression
虽然公式(4)中静电势方差参数在含氟高能致爆材料晶体密度预估公式中作用不明显,但将公式(4)中静电势方差替换为其他静电势参数的组合,可能会更好地体现含氟高能致爆材料体系内相互作用。由此课题组基于符号回归概念,开展含氟高能致爆材料的晶体密度预测研究。在本研究中,考虑到公式(2)以及(3)中静电势参数主要的运算方式为“加、减、乘、除”(分别对应于“add、sub、mul、div”等4个算符),
因此,在符号回归中基于上述4种运算符构建模型。如图 5所示,利用符号回归模型后得到的含氟高能致爆材料晶体密度预测公式的二叉树表示,其中每个叶节点都是变量或者常数,而内部节点是选定的运算符。
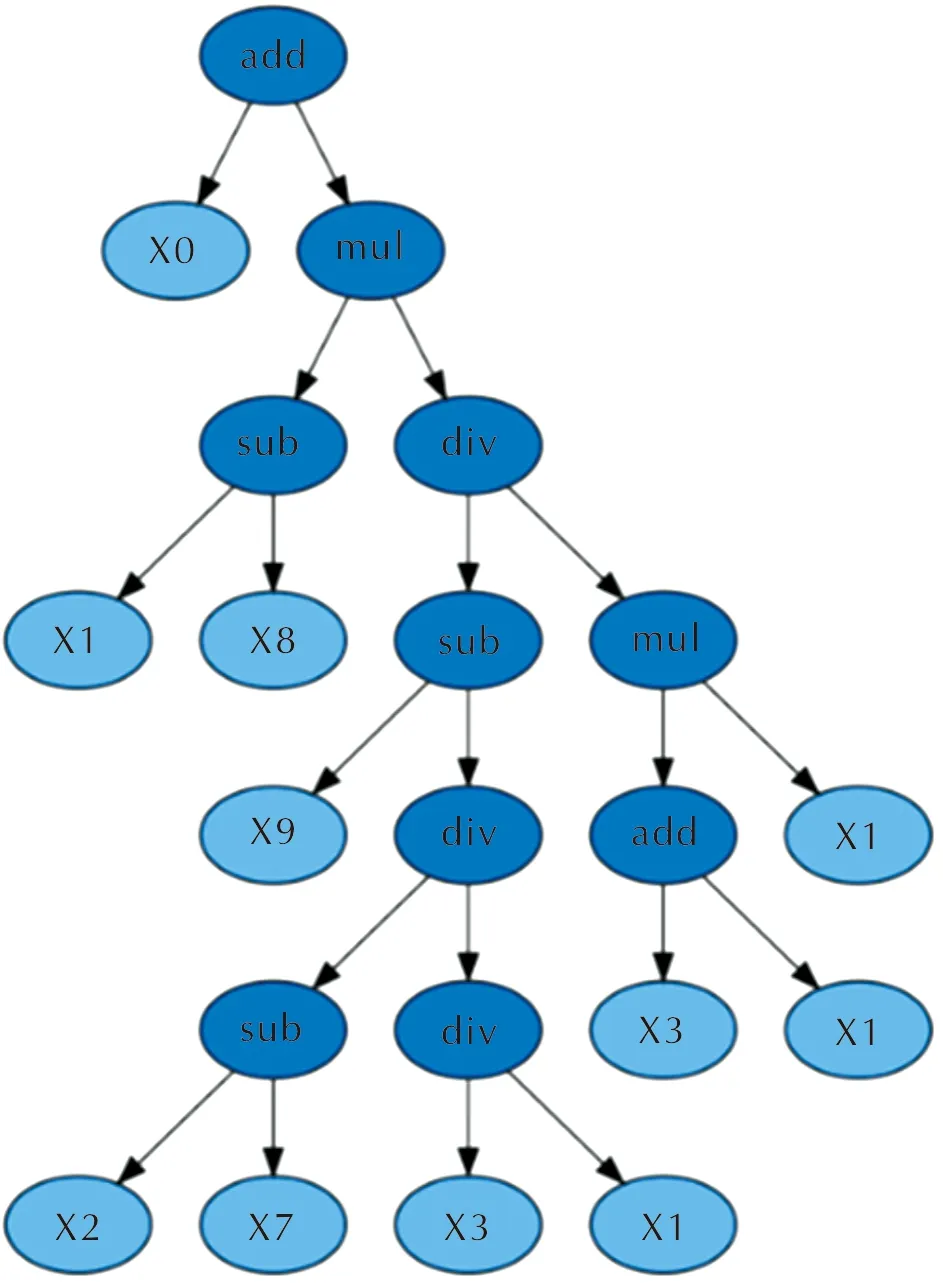
图5 含氟高能致爆材料晶体密度预测公式-二叉树表示Fig.5 Density prediction formula of fluorine-containing high energetic explosophoric materials-binary tree representation
根据图5的二叉树,可以得到其表达式,见公式(6):
(6)
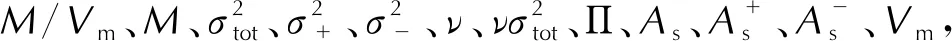
(7)
符号回归得到的密度预测公式,其R2=0.77,RMSE=0.045g/cm3,优于利用多元线性拟合方法修正的公式(4)。预测密度与实验晶体密度的分布如图 6所示,可以发现该公式弥补之前数个公式在预测实验密度低于1.75g/cm3的材料时高估密度以及预测实验密度高于2.00g/cm3的材料时低估密度的问题,表现在图中就是头尾的数据更加趋近于蓝色点线。如表2所示,相比于多元线性拟合方法,符号回归在降低预测均方根误差的同时,还提高了预测公式的综合表现。具体而言,一半以上材料的密度预测误差控制在0.03g/cm3以下,超过70%材料的预测误差低于0.05g/cm3。结合之前对于公式(2)、(3)和(4)的分析,公式(7)等号右侧第二项是由静电势方差、静电势偏差、0.001a.u电子密度等值面面积以及分子摩尔质量组合的新变量,该参数大致体现含氟高能致爆材料复杂的分子间相互作用并以数学表达式呈现,使得该公式能够弥补之前谈到的M/Vm对密度的高估问题,从而也就解释了为何该公式在密度预测时较优的表现。
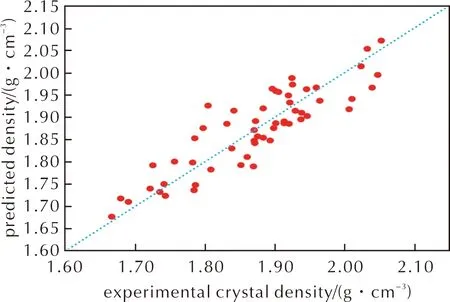
图6 符号回归方法改进后密度预测公式中预测密度与实验晶体密度关系Fig.6 The relationship between the predicted density and the experimental crystal density in the density prediction formula after the improvement of the symbolic regression method
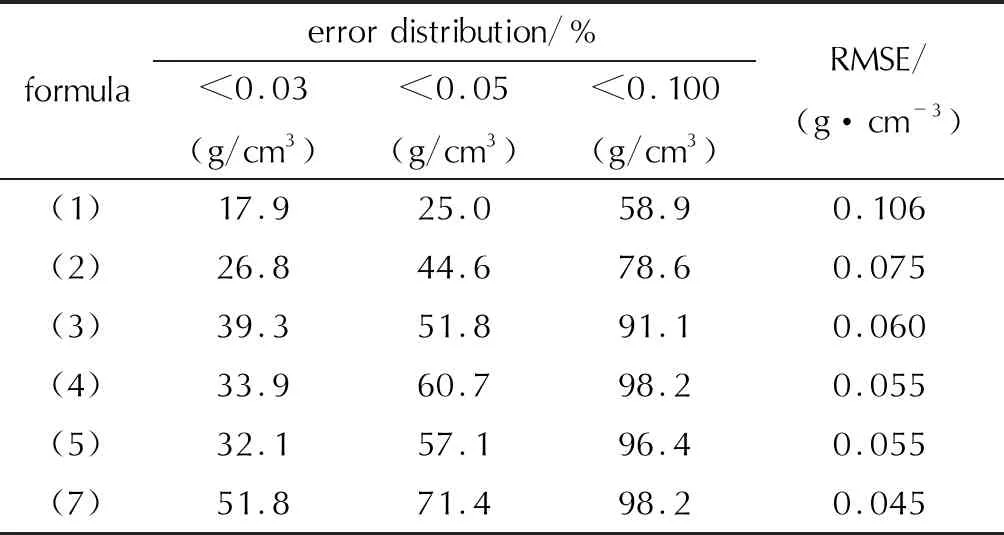
表2 各公式预测精度对比Table 2 Comparison of the prediction accuracy of different formulas
3 结 论
(1)基于分子体积方法,在Politzer的CHON晶体密度预测公式基础上,运用多元线性拟合得到了均方根误差为0.055g/cm3的修正公式。
(2)结合符号回归方法构建了适合含氟高能致爆材料晶体密度预测的公式,其均方根误差为0.045g/cm3,极大地提高了预测精度,其中有超过50%的化合物预测误差低于0.03g/cm3。
(3)符号回归构建的预测公式提高了新型含氟高能致爆材料晶体密度预测精度,有助于加速含氟高能致爆材料的设计。与此同时,符号回归方法还可以运用于其他研究体系,以破译材料性能间高维复杂的关系,构建出可理解的数学表达式,为材料设计提供思路。
猜你喜欢
广州化工(2022年19期)2022-11-09 11:30:46
广州化工(2022年18期)2022-10-22 10:27:00
小哥白尼(军事科学)(2022年4期)2022-07-08 07:03:38
意林彩版(2020年3期)2020-11-22 20:54:12
硅酸盐通报(2020年1期)2020-02-25 10:01:30
大众电视(蓝天下)(2018年8期)2018-10-26 01:00:40
能源(2017年5期)2017-07-06 09:25:55
西北工业大学学报(2015年4期)2016-01-19 03:31:58
海峡姐妹(2015年7期)2015-02-27 15:11:35
应用技术学报(2014年1期)2014-02-28 14:52:12
