
免疫检查点抑制剂治疗非小细胞肺癌耐药机制研究进展*
2023-01-14 07:22王怡鑫李凤何金涛陶婉君
肿瘤预防与治疗 2022年12期
王怡鑫,李凤,何金涛,陶婉君
610041 成都,四川省肿瘤医院·研究所,四川省癌症防治中心,电子科技大学 药学部(王怡鑫)、胸外科(何金涛);610054 成都,电子科技大学 医学院(李凤);610041 成都,四川大学 华西药学院(陶婉君)
免疫治疗主要由程序性死亡受体1(programmed cell death protein 1,PD-1)/程序性死亡配体1(programmed cell death ligand 1,PD-L1)抑制剂为代表,已在全球范围被批准用于治疗晚期非小细胞肺癌(non-small cell lung cancer,NSCLC)。PD-1属 于CD28家族成员,由Pdcd1基因编码,是一种单体糖蛋白,在胸腺细胞和在外周激活的CD4+和 CD8+ T细胞,B细胞、自然杀伤T 细胞、单核细胞、树突状细胞等均有表达[1]。PD-1有两个配体,即PD-L1和 程序性死亡配体2(programmed cell death ligand 2,PD-L2),PD-L2 较 PD-L1 对 PD-1 的亲和力高,但其表达水平低,故 PD-L1 显示出较为突出的调控作用[2]。除了与PD-1结合外,PD-L1还作用于CD80,而PD-L2是对排斥性导向分子B(repulsive guidance molecule B,RGMB)产 生 作 用。PD-L1与CD80结合可抑制T细胞,但PD-L2与RGMB结合则会促进T细胞的功能激活。也就是说,PD-L2对T细胞的活化既有促进作用又存在抑制作用。因此,抑制PD-L2可能无法达到促进T细胞活化的目的[3]。PD-L1 除表达于 T 细胞、B 细胞、树突状细胞等外,还高表达于黑色素瘤、肺癌、卵巢癌、淋巴瘤等多种实体肿瘤细胞表面[4-5]。实体肿瘤可以通过作用域PD-1/PD-L1轴,引起PD-L1过表达,诱导免疫抑制,从而抑制常规细胞毒性CD8+T细胞的攻击,防止肿瘤细胞溶解凋亡。因此,抑制PD-1/PD-L1途径会触发肿瘤抗原识别、增殖、浸润和细胞毒性CD8+T细胞的活化,从而产生抗肿瘤免疫反应[6]。目前,PD-1/PD-L1 抑制剂已经被应用于多种实体肿瘤,在临床上取得了较好的疗效,但该疗法仅对部分患者有效,有部分患者虽然PD-1/PD-L1 阳性表达,却对药物无反应或仅获得部分缓解,另外,越来越多起初对治疗有反应的患者最终疾病进展,表现为原发性或获得性耐药。
免疫介导的抗肿瘤活性依赖于复杂的动态机制,涉及肿瘤细胞、免疫细胞和信号介质之间的多种相互作用。对免疫治疗的效果和持续时间与这些不断发展的相互作用密切相关,而这些相互作用又受到选择性治疗压力的影响。对PD-1/PD-L1单抗治疗的耐药性可分为原发性耐药性和继发性(或获得性)耐药性[7]。原发性耐药通常定义为缺乏客观反应或肿瘤在治疗时间6个月内进展;而获得性耐药性将在观察到客观肿瘤反应或治疗时间超过6个月后进展。肿瘤内源性和外源性(或宿主)因素都可能导致原发性或继发性耐药[7]。
本文总结了最新现有研究,旨在阐明NSCLC对PD-1/PD-L1免疫检查点抑制剂耐药的机制,为NSCLC治疗方案的选择及免疫治疗的联合用药提供思路。
1 NSCLC PD-1/PD-L1免疫检查点抑制剂免疫治疗:原发性耐药机制
NSCLC对PD-1/PD-L1单抗的原发耐药性主要有两种(图1),另一种是肿瘤本身不被免疫系统识别,一种是适应性机制促进免疫逃逸,基于适应性机制产生的耐药[8]。各种宿主和/或肿瘤内在因素有助于原发耐药性。宿主因素包括肿瘤微环境(tumor micro-environment,TME)、内分泌和代谢因素、环境因素(如生态失调或抗生素或类固醇使用)和其他不可改变的因素(如年龄、慢性疾病或不利的宿主遗传因素)[9]。
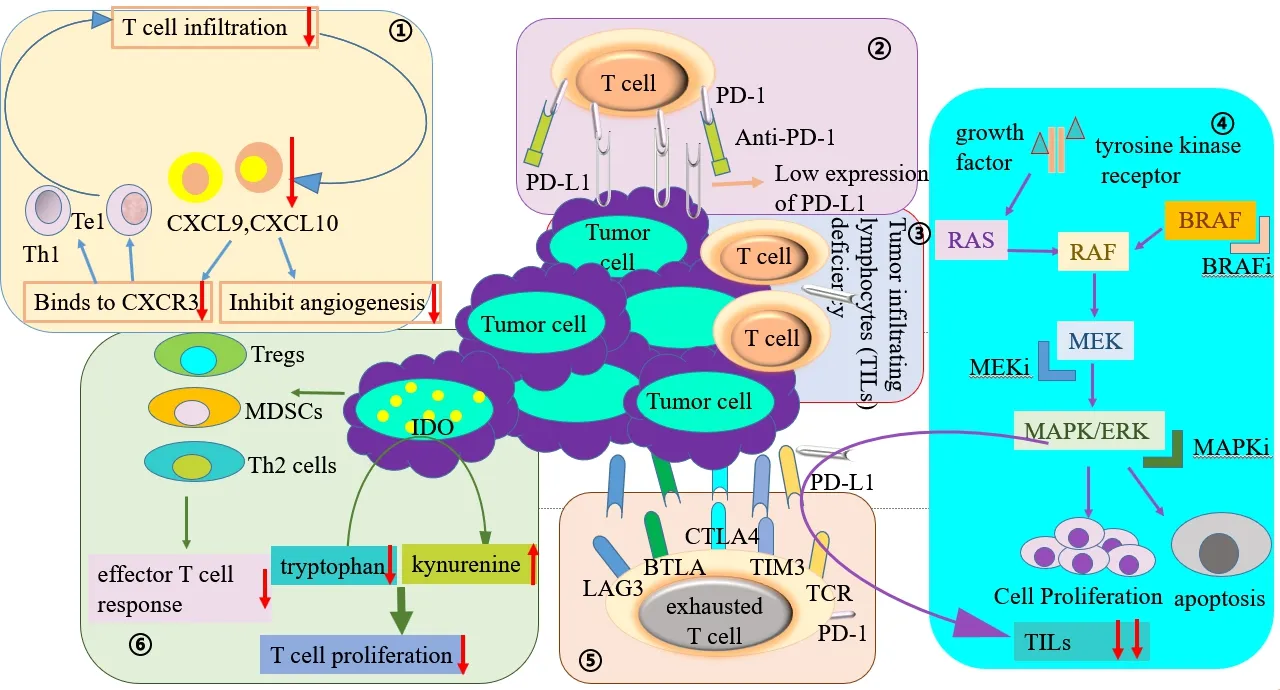
图1 NSCLC对 PD-1/PD-L1单抗的原发性耐药性主要机制Figure 1.Main Mechanisms of Primary Resistance to PD-1/PD-L1 Monoclonal Antibody in NSCLC
1.1 PD-L1表达异常及突变
肿瘤细胞表面的PD-L1表达异常上调和PD-L1缺乏都可能导致PD-1/PD-L1阻断治疗无效。PD-L1表达异常上调,转录因子阴阳1诱导的PD-L1表达上调触发含有NOD-、LRR-和pyrin结构域3的炎症小体,通过HSP70-TLR4信号传导促进肿瘤WNT5α表达,非规范WNT配体激活YAP途径诱导趋化因子受体2(C-X-C motif chemokine receptor 2,CXCR2)配体,而粒细胞样髓系来源免疫抑制性细胞的粒细胞亚群依靠CXCR2抑制T细胞功能[10]。
另外,有研究表明PD-L1的变异蛋白PD-L1v242和PD-L1v229(PD-L1v242和PD-L1v229是 两 种C端剪接变异的PD-L1)都可捕获PD-L1单克隆抗体,从而减弱了PD-L1与PD-L1结合的数量,证明了PD-L1剪接变体可以竞争结合PD-L1单抗,是产生耐药性的机制之一[11]。PD-L1与其受体PD-1之间的相互作用促进免疫逃逸并抑制T细胞功能,如果肿瘤细胞上的PD-L1缺乏,此时阻断PD-L1和PD-1则会增强NSCLC的免疫逃逸。因此,PD-L1或PD-1的 表 达 是 抗PD-1/PD-L1免 疫治疗有效的先决条件。JAK1/2中的功能丧失突变可能导致PD-L1表达缺乏而无法对IFN-γ作出反应,从而产生抗PD-1/PD-L1免疫治疗的原发性耐药性[12]。另外由于JAK1/2下游的干扰素γ受体途径控制着对T细胞具有化学吸引作用的趋化因子 的 表达,例如CXCL9、CXCL10和CXCL11,因此JAK1/2功能丧失还可能导致T细胞浸润的缺乏[13]。由于肿瘤中预先存在的T细胞是对抗PD-1治疗反应的必要条件,JAK1/2功能丧失导致PD-1/PD-L1单抗缺乏反应,不仅因为PD-L1不能反应性表达,而且还因为肿瘤细胞缺乏趋化因子而无法吸引T细胞。
1.2 T细胞本身的异常免疫
T细胞本身可能会发生对 PD-1/PD-L1 抑制剂的耐药性,即T细胞的异常免疫,主要表现形式为T细胞衰竭、T淋巴细胞浸润不足和T细胞功能障碍。T细胞衰竭是T细胞暴露于连续抗原信号并失去其效应器功能的状态,会促进原发耐药性[14]。有研究证明,高耗竭PD-L1的CD8+T细胞对PD-L1抑制剂的反应不佳[15]。T淋巴细胞浸润不足,治疗产生效果的关键前提是存在肿瘤浸润的细胞毒性T淋巴细胞(cytotoxic T lymphocyte,CTL)。即使PD-L1表达正常,缺乏T淋巴细胞浸润也可能导致PD-1/PD-L1阻断治疗无反应[16]。PTEN是一种脂质磷酸酶,可抑制激活途径的PI3K信号传导活性。据报道,PTEN的缺失可减少CD8+T细胞浸润到肿瘤中,并导致对PD-1阻断治疗的抵抗作用[17]。另一项研究报告说,肿瘤中WNT/β-连环蛋白途径的激活通过多种机制带来了非炎症环境,它作用于BATF3谱系的CD103DC,并诱导转录抑制剂激活转录因子3的表达,以减少Chemokine(C-C基序)配体4的产生,从而减少启动和浸润的CTL[18]。
1.3 其他信号通路因素导致免疫进化
RAF是有丝分裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)途 径 中 的 第 一 个 激酶,它磷酸化MEK,进而激活细胞外信号调节激酶(extracellular signal-regulatecl kinase,ERK)。ERK既激活胞质底物,又易位到细胞核,以参与刺激细胞增殖,存活,分化和细胞周期调节的各种基因的表达。已经广泛证明,MAPK信号通路在RAS介导的肿瘤发生中起着至关重要的作用[19-20]。MAPK途径介导多个细胞过程,如增殖、细胞凋亡和迁移,MAPK通路的畸变可能致癌,MAPK途径中的癌性突变通常主要影响细胞外信号调节激酶途径中的RAS和BRAF[21]。RAS-MAPK途径的改变抑制了NSCLC中T细胞的募集和浸润,促进肿瘤浸润淋巴细胞(tumor infiltrating lymphocytes,TILs)的耗尽和枯竭,从而导致PD-1/PD-L1阻断治疗耐药。在癌基因靶向治疗期间获得性iBRAF和iMAPK耐药性可以调节免疫微环境,促进TILs耗尽和衰竭,抑制PD-1/PD-L1阻断的治疗活性,有助于原发性耐药性。BRAF基因的突变在MAPK途径的顶部编码丝氨酸/苏氨酸激酶,在NSCLC中充当致癌驱动因素[22]。有研究证明circRNA C190是肺癌中多种促癌信号通路的介质致癌性,circRNA C190通过调节EGFR/ERK途径来激活MAPK/ERK依赖机制,C190的短暂和稳定过表达诱导ERK1/2磷酸化、增殖、体外迁移和异种移植肿瘤在体内生长[23-24]。
2 NSCLC PD-1/PD-L1免疫检查点抑制剂免疫治疗: 获得性耐药机制
获得性耐药性是指观察到客观肿瘤反应或治疗时间超过6个月后出现进展(图2)。靶向PD-1/PD-L1的免疫疗法仅阻断许多免疫检查点之一,但肿瘤仍有可能通过激活其他的抑制信号传导或升高PD-L1表达来逃避免疫消除。
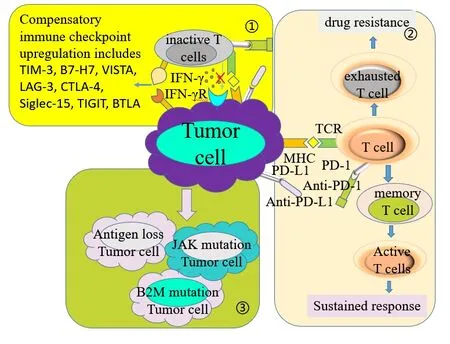
图2 继发性耐药机制Figure 2.Secondary Resistance Mechanisms
2.1 代偿性免疫检查点增多
免疫检查点代偿性增多,导致肿瘤细胞出现免疫逃逸或耗竭T细胞、抑制T细胞效应等,导致耐药。最新的代偿性免疫检查点研究情况见表1。

表1 代偿性免疫检查点激活机制及效果Table 1.Activation Mechanisms and Effects of Compensatory Immune Checkpoint Molecules
2.2 记忆T细胞分化失败
PD-1/PD-L1阻断剂可以重振功能低下的“疲惫”CD8+ T细胞(exhausted T cell,exT),恢复其抗肿瘤效应功能[33]。在免疫有效性发挥和抗原清除之后,一小部分效应T细胞分化为记忆T细胞,当重新识别出肿瘤抗原时,这些记忆T细胞将被迅速激活并增殖[34]。肿瘤浸润exT细胞的特征是染色质重塑和表观遗传修饰驱动的功能障碍增加,直到表观遗传功能障碍,染色质变得不可接近,对进一步的重塑和重振具有抵抗力[35]。如果肿瘤负荷仍然很高并重新焕发活力,exT细胞将无法根除肿瘤细胞,最终抗PD-1/PD-L1治疗使其重新衰竭并抵抗重振[36]。尽管缺乏针对NSCLC的研究,但对黑色素瘤患者的研究表明,肿瘤内记忆T细胞的扩增可能与抗PD-1/PD-L1治疗的疗效相关[37]。这表明记忆T细胞的产生受损可能导致PD-1/PD-L1阻断的疗效随着时间的推移而减弱,从而导致获得性耐药[38]。综上所述,这些研究表明,记忆T细胞的缺乏和重振耗尽的exT细胞可能导致对PD-1治疗的获得性耐药性[39]。
2.3 肿瘤的免疫编辑
耐药肿瘤中可能存在两种新抗原丢失机制,一种是通过免疫消除代表肿瘤细胞群一个子集的含有新抗原的肿瘤细胞,然后是剩余细胞的后续生长;另一种是通过在肿瘤细胞中获得一个或多个导致新抗原丢失的遗传事件,然后选择和扩增耐药克隆。该机制在抗PD-1/PD-L1治疗中不但能增强免疫力阻滞导致肿瘤进展,还会选择性逃避免疫系统导致肿瘤亚克隆,最终导致抗PD-1/PD-L1治疗的获得性耐药性[40]。有研究表明,肿瘤在体内暴露于产生IFNγ抗原特异性的CTL,会导致与DNA损伤反应和DNA编辑/修复基因表达调节相关的拷贝数改变[41]。因此CTL和IFN-γ免疫编辑可能是增强遗传不稳定性的机制之一,通过遗传进化而改变其免疫抵抗力,对PD-1/PD-L1阻断治疗产生获得性耐药性。一项在NSCLC患者对抗PD-1/PD-L1 和/或抗CTLA-4抗体的免疫检查点抑制最初反应后,出现获得性耐药期间肿瘤新抗原的演变的研究中发现,在具有获得性耐药性的肿瘤细胞中检测到7至18种推定的突变相关新抗原的丢失,这是基因组改变的结果导致新抗原丢失的免疫编辑机制可能导致肿瘤细胞获得性PD-1/PD-L1耐药性[42]。B2M是人类白细胞Ⅰ类抗原(human leukocyte antigen I,HLA-I)类复合物的重要组成部分,肿瘤B2M缺乏症(或有缺陷的细胞表面HLA-I表达)与免疫逃逸有关[43]。由ICI耐药肿瘤产生的3/5异种移植物表现出B2M下调,其中两个也具有丢失或几乎无法检测到的细胞表面HLA-I类表达水平,获得性B2M纯合子丢失导致肿瘤中缺乏细胞表面HLA-I类表达和匹配的患者来源异种移植物,获得性B2M突变导致的HLA-I类APM破坏可以介导NSCLC免疫逃逸[44]。有研究表明JAK1或JAK2的失活可能对癌细胞特别有利,编码干扰素受体相关的JAK1/2的基因中显示出与抗性相关的功能丧失突变,同时缺失野生型等位基因,JAK1/2截断突变导致对干扰素γ缺乏反应,导致其对癌细胞的抗增殖作用不敏感[45]。JAK1/2对于介导IFN相关信号传导至关重要,肿瘤细胞对T细胞来源的IFN变得明显不敏感,这意味着肿瘤细胞对T细胞抑制作用不敏感。
3 总结与展望
迄今为止,以PD-1/PD-L1阻断治疗为代表的免疫治疗,处于抗肿瘤临床治疗的前沿,已经使许多患者受益。然而,这种阻断免疫疗法的原发性和获得性耐药性的存在限制了其疗效。为了使NSCLC使用PD-1/PD-L1抑制剂进行免疫治疗产生疗效,基于目前对于耐药机制的基础研究,需要满足六个条件:(1)足够免疫原性肿瘤;(2)T细胞对NSCLC肿瘤微环境充分活化和浸润;(3)肿瘤依赖PD-1/PD-L1轴来逃避抗肿瘤免疫应答;(4)衰竭的肿瘤特异性CD8+T细胞充分重振;(5)肿瘤细胞表面的MHC上显示这些T细胞特异性的肿瘤抗原或肿瘤生长所必需的蛋白质;(6)分泌出长效效应T细胞及足够的记忆T细胞(持续反应)。
因此,NSCLC中的PD-1/PD-L1阻断免疫治疗抗肿瘤活性可以通过以下方式增强:(1)通过个性化疫苗、CAR-T、TILs、放疗/化疗以增加新抗原识别和T细胞活化,促进T细胞有效启动;(2)靶向TIM-3、B7-H7、VISTA、LAG-3、CTLA-4、Siglec-15、TIGIT、BTLA活性,达到去除共抑制信号的目的,避免替代免疫检查点上调,以支持阻断PD-1/PD-L1轴后免疫效应细胞功能;(3)缓解缺氧状态,靶向IDO、VEGF、CDK4/6、细胞因子、致癌途径、转录调节剂,调节肿瘤微环境以增强效应T细胞浸润;(4)调节肠道微生物群,促进TME塑造抗肿瘤有利的影响。
目前已有相关联合策略进行临床试验。TIGIT是目前NSCLC中唯一可以获得随机临床数据的新靶点,CITYSCAPE是一项临床Ⅱ期试验,该试验招募了新诊断的局部晚期或转移性NSCLC患者,这些患者在至少1%的肿瘤细胞中表达PD-L1并且没有EGFR或ALK改 变。TIGIT抑 制 剂tilagorumab与PD-L1抑制剂atezolizumab联合治疗组取得了统计学上显着的结果[46]。Ⅲ期SKYSCRAPER-01(NCT04294810)研究评估了在atezolizumab联合tilagopab作为局部晚期或转移性NSCLC患者的一线治疗,目前还没有达到其无进展生存期的共同主要终点,预计在2025年2月全部完成(2022年7月31日访问clinicaltrials.gov)。由于肿瘤的异质性和可塑性,需要对癌症患者进行识别分类制定个体化治疗方案开展精准治疗。根据患者的自然病程,细胞和分子肿瘤特征以及其免疫反应能力来设计治疗策略。因此,对NSCLC的细胞和分子组成表征研究以及确定对癌症免疫治疗的反应率的预测性生物标志物的鉴定,对提高临床使用PD-1/PD-L1抑制剂的治疗效果有极大帮助。
作者声明:本文全部作者对于研究和撰写的论文出现的不端行为承担相应责任;并承诺论文中涉及的原始图片、数据资料等已按照有关规定保存,可接受核查。
学术不端:本文在初审、返修及出版前均通过中国知网(CNKI)科技期刊学术不端文献检测系统的学术不端检测。
同行评议:经同行专家双盲外审,达到刊发要求。
利益冲突:所有作者均声明不存在利益冲突。
文章版权:本文出版前已与全体作者签署了论文授权书等协议。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
计算机系统应用(2022年4期)2022-05-10
天津医科大学学报(2021年4期)2021-08-21
临床肝胆病杂志(2020年2期)2020-12-14
中国生殖健康(2020年7期)2020-12-10
天津医科大学学报(2019年3期)2019-08-13
中国中医急症(2019年10期)2019-05-21
新西部下半月(2018年7期)2018-10-20
计算机应用(2017年1期)2017-04-17
上海医药(2017年1期)2017-03-01
