
密度泛函理论计算氢氧化镁与纤维二糖和重金属离子相互作用
2023-01-09 12:34王新宇潘清江
黑龙江大学自然科学学报 2022年3期
王新宇, 潘清江
(黑龙江大学 化学化工与材料学院 功能无机材料化学教育部重点实验室,哈尔滨150080)
0 引 言
人类社会和工业生产的高速发展已经引发诸多环境问题,如水污染防治已成为当今迫切需要解决的难题。为从废水中去除高毒性的Cd等重金属,以及实现放射性核废料的长期安全处置,人们已开发很多材料并尝试多种去污策略[1-2]。如采用二维层状Mg(OH)2材料对有污染的离子进行置换、吸附等反应,从而达到净化环境的目的。Mg(OH)2是一种来源广泛的无机材料,为层状结构,属于六方晶系,具有毒性低、环保、成本低等优点。目前,报道的合成方法有溶胶凝胶法、水热法和溶剂热法等,并已得到针状、棒状、片状和花状等多种形貌的样品[3]。
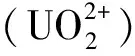
本文通过第一性原理计算研究Mg(OH)2材料的结构,并探索其对富含羟基的纤维二糖和有毒重金属离子(镉和铀)的吸附界面作用,以期对新纳米复合材料合成以及开发其在环境科学和能源领域的应用提供理论支持。
1 计算方法
采用VASP程序[8]进行基于平面波基组的第一性原理计算,使用了广义梯度近似(GGA)的Perdew-burke-ernzerhof(PBE)泛函[9]。为了准确描述材料界面的弱相互作用,计算中引入Gramme的D2色散校正[10]。电子结构的计算采用杂化泛函Heyd-Scuseria-Ernzerhof(HSE06)[11],截断能为550 eV,能量和力分别收敛于1×10-5eV和0.1 eV·nm-1。采用Monkhorst-Pack方法拟合Brillouin区域,其中k点设置为1× 1× 1。为了模拟Mg(OH)2的体相特性,仅优化表层原子,而固定内层原子。
底物Mg(OH)2(001)晶面的超胞大小为3×3×1,真空层厚度为1.5 nm。所有吸附质在计算中均做全优化处理。选择的第一种吸附质是纤维二糖(标记为Cel),分子式为C12H22O10。因为纤维二糖结构简单且富含羟基,可用于模拟生物质材料和多种工业有毒染料。建立了1~5层的Mg(OH)2基底模型,命名为MH_n(n= 1~5),复合纤维二糖以后命名为Cel-MH_n。计算和分析表明,三层Mg(OH)2可模拟底物的各种行为。
选择的第二种吸附质是铀酰离子。根据吸附剂的合成条件,铀酰在赤道平面上与水和羟基配位。结合之前的报道[12-14],选择赤道方向五配位的铀酰离子体系,分子式[(UO2)(H2O)5-n(OH)n]2-n(n= 0~3)。用以下公式计算复合材料的吸附能(EAds):
EAds=ECom-ESub-EAdsorbate
(1)
式中ECom、ESub和EAdsorbate分别对应优化的复合物、基质和吸附质体系的能量,EAds越负,表示吸附或界面相互作用越强。
还尝试用Mg(OH)2去除镉离子。根据离子交换反应[15],置换能(ERep)计算公式为:
(2)
式中涉及体系均为优化几何下的能量。
2 结果与讨论
2.1 Mg(OH)2结构对纤维二糖的吸附作用
首先,将第一种吸附质,纤维二糖置于底物的表面上,确定了最具代表性的三层Mg(OH)2-纤维二糖复合物(标记为Cel-MH_3),优化的稳定结构如图1(a)所示。在图1(b)中,对参与界面反应的原子标记并编号。纤维二糖的H原子和O原子用C标记,用M标记Mg(OH)2的原子。复合物Cel-MH_3界面处的键长和键角如表1所示。界面处的HC—OM键长为0.150~0.218 nm。计算得到的OC—HC—OM键角为152.7°~177.4°,符合氢键特征。相比之下,OC—HM距离为0.194~0.208 nm,OC—HM—OM键角相对较小,为138.8°~162.5°。因此HC和OM之间存在较强的界面氢键。

图1 (a)PBE+D2优化的Cel-MH_3的结构和(b)H和O原子的编号,其中纤维二糖和Mg(OH)2分别标记为C和M
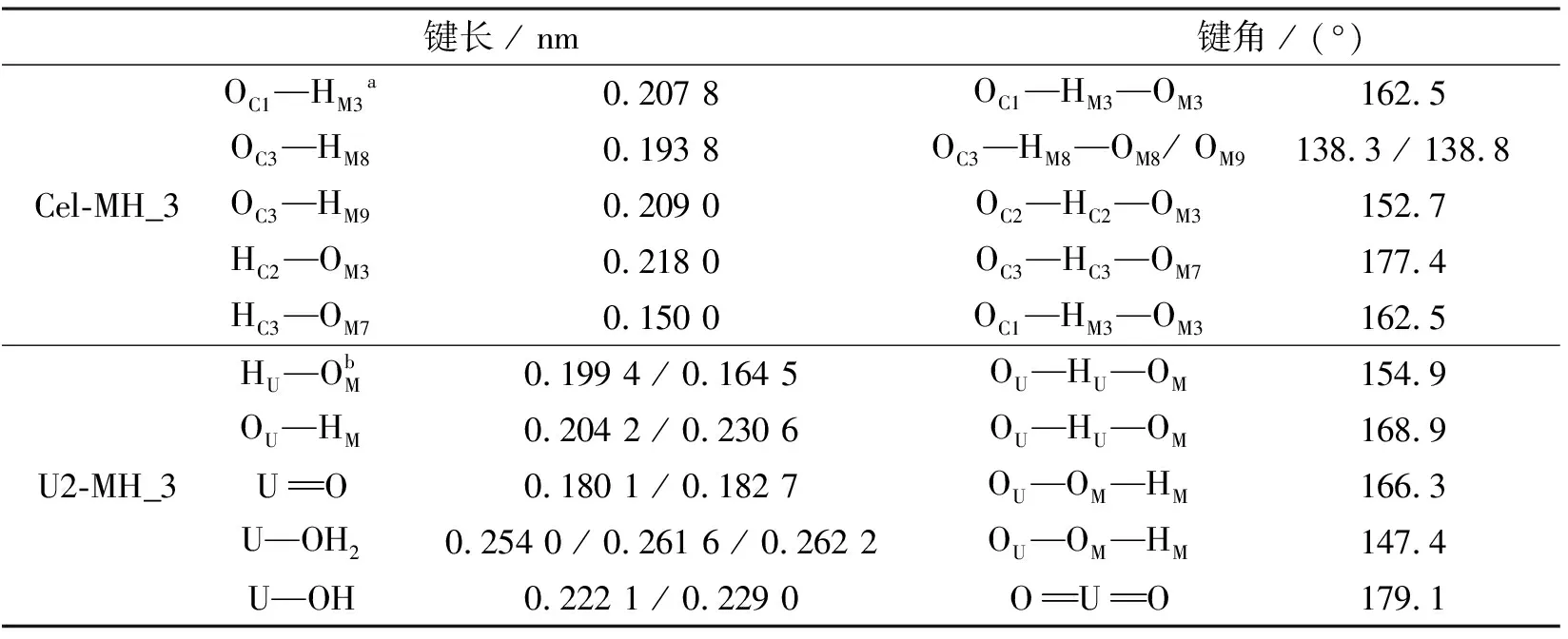
表1 优化Cel-MH_3和U2-MH_3的几何参数
根据计算方法中的式(1),计算Mg(OH)2对纤维二糖的吸附能(EAds),具体数值如表2所示。计算的复合物Cel-MH_n(n= 1~5)的EAds为-0.80~-1.06 eV。由于纤维二糖末端的三个—OH基团参与了界面相互作用,为了使结果变得具有合理性和普适性,平均吸附能(EAds(av))为-0.27~-0.35 eV,它的强度完全符合报道的氢键强度范围[16]。结合上述优化几何参数的结果,验证了界面之间的相互作用本质是氢键。复合材料的基底Mg(OH)2的吸附能(绝对值)在1~3层时增大,在第4层时减小,在第5层时增大。这显示出振荡趋势,与其他材料的计算结果[17]一致。可推导出EAds和EAds(av)分别收敛于-0.91和-0.30 eV。比较发现,三层Mg(OH)2足以代表大的体相底物。

表2 计算的Mg(OH)2对纤维二糖和铀酰的EAds和EAds(av)(eV)
2.2 Mg(OH)2-纤维二糖的电荷和电子性质
用电荷密度差分(CDD)图表征了复合材料Cel-MH_3的界面相互作用。如图2所示,黄色区域为电子积聚,青色区域表示电子缺失。可以注意到,电荷在Mg(OH)2和纤维二糖之间的界面重新排布,表明它们之间发生了化学耦合。尽管层数发生变化,但是电荷积聚都发生在界面的H原子和O原子之间。总之,复合材料的界面中发生的化学耦合是氢键本质。

图2 计算的Cel-MH_3、U2-MH_3和Cd@MH_3的CDD图,其中青色区域代表电子损失,黄色区域为电子积聚
电荷(Bader)计算结果表明,纤维二糖部分始终显示负电性,而Mg(OH)2部分则显示正电性。随着层数的变化,电子从Mg(OH)2基底转移到纤维二糖中,转移量为0.048~0.106。这个小的电荷转移(CT)量也可以证明界面相互作用是氢键性质,与上述CDD和几何分析的结论相一致。随着层数的增加,CT量也像上述计算的EAds值一样呈现振荡趋势。它们之间大致是正相关的。
为进一步研究Mg(OH)2吸附纤维二糖的行为,使用HSE06杂化泛函和Grimme的D2色散校正得到了每个复合物Cel-MH_n(n=1~5)的DOS图,如图3所示。当层数从1变化到5时,复合材料的带隙分别为4.38、4.59、4.58、4.10和4.65 eV。它们表现出振荡趋势,与上述的EAds值呈正相关。为了便于观察,将CBM附近的峰放大,如图3右侧所示。把Mg(OH)2基底分成两部分描述:一部分是与纤维二糖直接接触的表层Mg(OH)2,另一部分则是内层Mg(OH)2。VBM轨道的贡献来自于表层Mg(OH)2,组分中的纤维二糖成分主要贡献于CBM轨道,此外对于n=2、3和5时的Cel-MH_n复合物,CBMs中也掺杂一些内层Mg(OH)2的贡献。

图3 在HSE06+D2理论级别计算的复合物Cel-MH_n的DOS图
2.3 Mg(OH)2处置重金属镉和铀酰离子
二维Mg(OH)2材料通过阳离子交换机理去除以Cd为代表的重金属离子以净化工业废水[4, 15]。采用Cd2+取代Mg(OH)2表面的Mg2+来构建模型。上述研究证实了三层Mg(OH)2(标记为MH_3)是一种良好的基底模型。因此,优化得到Cd置换MH_3的模型(标记为Cd@MH_3)如图4所示。根据式(2)计算得到置换能(ERep)为-0.79 eV,这表明Mg(OH)2去除废水中的Cd2+在热力学上是有利的。计算得到的Bader电荷表明,Mg带1.67个正电荷,比带1.23个正电荷的Cd更接近预期的+II氧化态。这些计算值与它们的电负性(Mg为1.31,Cd为1.69)相一致。从CDD图(图2)可以看出,在Cd原子周围电荷重新排布,电荷分布表明Cd与O之间具有化学键合特性。

图4 优化的Cd@MH_3复合物以及其他反应试剂的结构
铀酰离子易溶于水,其处理、存储放置和运输都可能会造成严重的环境污染[12-14]。由于Mg(OH)2纳米材料具有优异的吸附性能,可应用于放射性有毒铀酰固化、污水治理和海水提铀[7]。本文选择了H2O或—OH以五配位的形式构建铀酰模型,使用PBE+U方法计算含铀材料,Ueff参数值设为2 eV[18]。同时,计算中包含色散校正,PBE优化得到了4种稳定的结构[(UO2)(H2O)5-n(OH)n]2-n(n= 0~3,标记为Un),如图5所示。当n= 2时,2个—OH有两种排列方式,产生异构体[(UO2)(H2O)2(OH)(H2O)(OH)]和[(UO2)(H2O)3(OH)2]。优化后,后者转化为[(UO2)(H2O)2(OH)2]·(H2O),其中1个H2O位于第二配位壳层。当n= 4 和5时,也发生H2O或—OH跑到第二配位壳层的情况,不满足赤道五配位模式。因此,不考虑这些吸附质。
但是,当把铀酰物种U0和U1放到MH_3表面时,并没有得到能量上有利的结构,这从静电的角度来看是可以理解的。Bader电荷计算表明,MH_3表面暴露的H原子带正电荷,U0和U1分别带+2和+1价的电荷,所以MH_3与U0和U1之间存在静电排斥作用,导致得不到能量有利的结构。
中性的U2是通过1个—OH和1个H2O与MH_3结合,而负电性的U3则通过2个—OH的方式结合,它们的吸附方式是不同的。图5为优化后的U2-MH_3和U3-MH_3的稳定结构,计算得到其EAds值分别为-1.64和1.32 eV,这说明Mg(OH)2容易吸附水中的U2型铀酰,对其具有较强的吸附能力。如表1所示,计算界面处有4个H…O键,其间距为0.165~0.231 nm,所以每个键的平均吸附能是-0.41 eV,属于氢键强度范围。U2-MH_3的CDD(图2)显示,铀酰上的-yl氧原子、水基团的H以及整个—OH都有电荷向Mg(OH)2表面的—OH转移。MH_3向U2的CT量为0.15,结合上述CDD可证明界面之间是氢键作用本质。因此,可判断Mg(OH)2在弱碱性条件下吸附铀酰的吸附机理是一种借助界面氢键的外配位壳层机理。

图5 优化的吸附质[(UO2)(H2O)5-n(OH)n]2-n(Un,n = 0~3)和复合物Un-MH_3(n = 2和3)的结构
3 结 论
对Mg(OH)2和吸附纤维二糖复合材料的计算显示,吸附质的3个—OH参与了界面相互作用。得到的平均吸附能为-0.27~-0.35 eV,在典型的氢键强度范围内,指认界面间作用为氢键本质。随着Mg(OH)2层数的增加,计算的吸附能、基底→吸附质的CT量和Eg表现出振荡趋势,且这些量之间存在近似的正相关性。采用三层Mg(OH)2模型与Cd2+反应,计算得到的置换能为-0.79 eV,表明Mg(OH)2去除废水中Cd2+在热力学上是可行的。计算表明,Mg(OH)2更容易吸附由3个H2O和2个—OH配位的铀酰离子,其中吸附质呈现电中性,计算的吸附能为-1.64 eV。在弱碱性条件下,Mg(OH)2材料吸附铀酰主要为界面氢键作用,遵循外壳层吸附机理。本研究为探索界面化学、设计和开发新材料以及挖掘其潜在应用奠定了理论基础。
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
波谱学杂志(2021年3期)2021-09-07
——以高中化学“氢键”的教学为例
教学月刊(中学版)(2020年13期)2020-12-29
杭州化工(2020年2期)2020-01-16
中成药(2019年12期)2020-01-04
人工晶体学报(2019年5期)2019-06-18
当代陕西(2019年6期)2019-04-17
分析化学(2018年2期)2018-03-02
中成药(2017年5期)2017-06-13
中学化学(2015年12期)2016-01-19
